|
Студопедия КАТЕГОРИИ: АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция |
Отбор первичной пробы твердых веществТвердые вещества могут быть порошкообразными (сыпучими) и кусковыми. Чем крупнее куски материала, тем больше его неоднородность и тем сложнее взять среднюю пробу. Способ отбора пробы зависит не только от состояния материала (порошкообразный, кусковой), но и от того, поступает материал без упаковки или в таре (мешки, ящики, бочки, банки). При этом надо учитывать степень его однородности и условия хранения. Вследствие небрежного хранения материала он увлажняется, загрязняется. Отбор первичной пробы сыпучих материалов,поступивших в таре, берут приблизительно в равных количествах из разных мест (снизу, сверху, из середины, с боков) с помощью специального приспособления— щупа. Он представляет собой железный или медный узкий желоб, заостренный снизу, с рукояткой для удобства пользования (рис. 7, а, б). Щуп вкручивают в глубину материала и вещество насыпается в желоб. Поскольку материал частично сползает по желобу, нижние слои не попадают в состав пробы, используютщуп конструкции Говальского (рис. 7, б), позволяющий отбирать пробу более точно. На его конце имеется пластинка-клапан, вращающийся перпендикулярно оси щупа. Ручка 2 подвижна и при движении вперед фиксирует клапан так, что щуп остается открытым и наполняется веществом. Чтобы вытащить щуп из материала, ручку выдвигают назад, при этом клапан освобождается и при движении щупа материал давит на клапан, который закрывается и проба остается внутри щупа.
Если сыпучий материал рассыпан относительно тонким слоем (до 1 м) на большей площади ;(в вагоне), то следует брать пробу в нескольких точках, расположенных в шахматном порядке. Пробу вязких, пастообразных масс надо отбирать по всей толщине массы сверху до дна.  Отбор первичной пробы кусковых материаловнаиболее труден. Состав кусков может резко отличаться. При отборе пробы необходимо сохранять в ней такое же соотношение между крупными кусками и мелкими, как в исходном материале. Наиболее точно отбирается проба, если весь материал измельчить. Если по технологическому процессу для переработки исходное сырье подлежит измельчению, то целесообразно брать пробу после измельчения. Обработка отобранной первичной пробы.Первичная средняя проба твердых материалов может иметь массу в несколько килограммов, тогда как для химического анализа (лабораторная проба), как правило, требуется несколько граммов. Отсюда следует необходимость сокращения пробы без изменения ее состава. Это достигается разделкой первичной пробы такими операциями, как измельчение, перемешивание и сокращение. Измельчение осуществляют вручную с помощью ступки или трамбовки на стальной плите. Ручное дробление трудоемко, его применяют в тех случаях, когда редко отбирают пробы небольшой массы. Для регулярной переработки большого количества проб применяют специальные дробильные приспособления. Перемешивание небольших масс пробы осуществляют при помощи совка вручную. В случае значительной массы пробы ее насыпают лопатой в виде конуса, затем перекидывают на новое место и опять собирают в виде конуса. Сокращение производят по определенным правилам, чтобы лабораторная проба по составу соответствовала первичной. Наиболее надежным способом сокращения пробы является способ кольца и конуса с последующим квартованием. Всю пробу высыпают на ровную поверхность (железный лист или пол), распределяют в виде кольца треугольного сечения (рис. 8) и затем собирают в конус в центре этого кольца. В вершину конуса вертикально вставляют острый край деревянной дощечки и поворачивая ее, как показано на рис. 9, развертывают конус в диск Подобную операцию повторяют 2—3 раза, после чего диск квар-туют, т. е. делят на четыре равных сектора двумя перпендикулярными бороздами, проходящими через центр. Два противоположных сектора отбрасывают, а два оставшихся перемешивают и подвергают дальнейшему сокращению по описанной методике до тех пор, пока не останется масса, требуемая по ГОСТу Отбор лабораторных пробпроводят способом вычерпыванияпробу, разровненную в виде круга, делят шпателем на ряд взаимно перпендикулярных рядов и отбирают лабораторную пробу в шахматном порядке из середины образовавшихся квадратов на всю глубину слоя вещества.
Отбор проб жидкостей Среднюю пробу жидкости берут специальным пробоотборником, конструкция которого зависит от вида анализируемой жидкости. Методы отбора проб и количество отбираемой жидкости в каждом конкретном случае определяются стандартами и техническими условиями. Отбор проб из больших резервуаровзависит от однородности или неоднородности жидкости, подлежащей анализу. Если жидкость однородна, то достаточно зачерпнуть необходимое ее количество в любом месте, чтобы получить среднюю пробу. Если же жидкость неоднородна (осадок, муть), среднюю пробу составляют из проб, отобранных на разных уровнях жидкости. Принято отбирать три или пять проб в зависимости от высоты столба жидкости. В этом случае измеряют толщину каждого слоя, а затем отбирают пробу пропорционально объему. Так, при отборе трех проб одну из них берут на 0,5 м от поверхности, вторую—на расстоянии 0,5 м от дна и третью — на середине. Из неглубоких резервуаров пробы берут при помощи стеклянных трубок с оттянутыми концами, из глубоких — специальными пробоотборниками. Отбор проб из мелкой тары(бидоны, бутыли, железные бочки и т. л.) берут от определенного процента мест поступившей партии. Жидкость в сосуде перемешивают и отбирают пробу при помощи длинной, суженной на конце стеклянной трубки, медленно погружая ее в жидкость в вертикальном положении. Затем, зажав верхнее отверстие трубки, ее вынимают. Так отбирают пробы из всех предусмотренных тарных мест и смешивают в приемнике. Количество отбираемой жидкости — не менее 1 л. Полученную среднюю пробу разливают в две сухие чистые склянки. Одна из них поступает на анализ, другая сохраняется на случай арбитражного анализа. Отбор пробы промежуточных продуктов из аппаратов и трубопроводов проводят при постоянном контроле за ходом технологического процесса. Отбирают индивидуальные пробы, которые сейчас же анализируют взятие проб из аппаратов производят через вентили или специально установленные краны. Пробу жидкости, текущей по трубопроводу, отбирают через пробоотборный кран специальной конструкции (рис. 10), соединенный с несколькими трубками, загнутые концы которых направлены отверстиями навстречу текущей жидкости. Трубки позволяют отбирать пробу в разных слоях жидкости.
Лекция 4 ГЛАВА 4 ОПРЕДЕЛЕНИЕ ФИЗИЧЕСКИХ ПОКАЗАТЕЛЕЙ МЕТОДАМИ, ПРИМЕНЯЕМЫМИ В ТЕХНИЧЕСКОМ АНАЛИЗЕ
ПЛОТНОСТЬ Различают абсолютную и относительную плотность. Абсолютной плотностьюр (кг/м3) называют массу вещества, содержащуюся в единице объема. За единицу принимают массу 1 м3 чистой воды при 4°С. Относительной плотностьюр420Называют отношение массы вещества к массе чистой воды при 4°Св одинаковом объеме ее определяют, как правило, при 20°С и относят к плотности воды при 4°С. Относительная плотность — безразмерная величина. Если, например, плотность воды измерена не при 4°С, то полученное значение относительной плотности жидкости необходимо пересчитать, умножив его на отношение плотности водыпри той же температуре к ее плотности при 4°С. Например, надо пересчитать относительную плотность жидкости, определенную при 20 С, на плотность воды
при 4°С:
Плотность вещества можно определить при любой температуре, пересчитаее на р420 по формуле
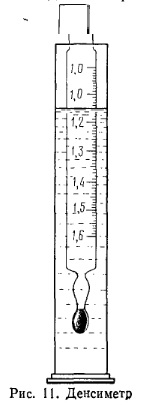
где р4t — плотность вещества при температуре испытания; t— температура испытания; α — поправочный температурный коэффициент (см. табл. 2приложения). Плотность характеризует идентичность, чистоту и концентрацию вещества. Для многих веществ (спирт, глицерин, формалин и др.) установлена зависимость между плотностью и концентрацией. Зная плотность вещества, по таблице можно найти его концентрацию (см. приложение) и, наоборот, по известной концентрации по этой же таблице находят плотность вещества. Экспериментально плотность определяют денсиметром или гидростатическим взвешиванием. Оба эти способа основаны на законе Архимеда. При определении относительной плотности можно использовать пикнометр. Плотность, определяемую пикнометром или гидростатическим взвешиванием, называют видимой, так как взвешивание проводят в воздухе, плотность которого не учитывают. Для более точных измерений необходимо ввести поправку на потерю массы в воздухе по формуле
где pист и p1 — соответственно истинная и видимая плотности; 0,99823 — плотность воды при 20°С; 0,0012 — плотность воздуха. Работа 1. Определение плотности жидкости с помощью денсиметра (ГОСТ 18995.1—73). Денсиметр (ареометр) представляет собой стеклянный цилиндрический сосуд, нижняя часть которого заканчивается шаром, заполненным свинцовой дробью (рис. 11). На цилиндрической части денсиметра нанесена шкала с. делениями, обозначающими плотности жидкостей, в которые погружают денсиметр, и температуру, при которой следует производить определение. Градуировку денсиметров производят при 20°С и относят к плотности воды при 4°С. Для повышения точности измерения и удобства пользования изготовляют набор денсиметров, шкалы которых охватывают определенный диапазон плотностей. Техника определения. В чистый цилиндр внутреннего диаметра не менее 5 см наливают испытуемую жидкость. Стараясь не задеть стенки цилиндра, в жидкость медленно вводят чистый и сухой денсиметр; держа его за верхний конец, ожидают (2—3 мин), чтобы денсиметр пришел в равновесие. Необходимо, чтобы при этом он не касался ни дна, ни стенок цилиндра. Отсчитывают деление на денсиметре по верхнему краю мениска и замеряют температуру анализируемой жидкости термометром, опущенным в цилиндр. Если температура испытуемого продуктаотличается от 20°С, а ареометр градуирован на р4, то вводят поправку на найденную плотность (см. табл. 2 приложения). Допустимое расхождение между двумя параллельными определениями не должно превышать 0,001. Денсиметры содержат в сухом чистом виде, каждый раз после использования его надо промыть водой или соответствующим растворителем, вытереть и положить в набор.
Работа 2. Определение плотности при помощи пикнометра (ГОСТ 18995.1—73).Пикнометр представляет собой стеклянный сосуд с кольцевой меткой на шейке (рис. 12), емкостью от 1 до 100 мл (для работы наиболее удобны пикнометры емкостью 25— 50 мл). Техника определения. Перед определением пикнометр тщательно моют и высушивают в сушильном шкафу при 100°С. Затем его взвешивают на аналитических весах с точностью до 0,0002 г, наполняют дистиллированной водой чуть выше метки, помещают в термостат и выдерживают при 20°С в течение 15— 20 мин. Когда жидкость в пикнометре примет температуру термостата, жгутиками фильтровальной бумаги отбирают избыток воды до уровня метки на шейке. При определении плотности темных жидкостей уровень их в пикнометре устанавливают по верхнему мениску, а для светлых жидкостей — по нижнему; соответственно и уровень воды устанавливают по верхнему или нижнему мениску. Пикнометр закрывают пробкой, вынимают из термостата, тщательно вытирают снаружи фильтровальной бумагой и взвешивают. Находят объем пикнометра по формуле
Воду из пикнометра выливают и высушивают его в сушильном шкафу. Затем наливают в пикнометр испытуемую жидкость немного выше метки, помещают в термостат при 20°С и производят те же операции, что и с водой вплоть до взвешивания.
Плотность р420 испытуемого вещества находят по формуле
Работа 3. Определение плотности гидростатическим взвешиванием (ГОСТ 15139—69).Для измерения плотности отформованного изделия можно использовать стандартные образцы в виде брусков размером 10X15x120 мм или вырубать образцы размером 20X20X2 мм. Допускается испытание образцов произвольных размеров или целых изделий при условии, что их объем не менее 1 см3. Для проведения испытаний можно использовать аналитические весы, специально приспособленные для гидростатического взвешивания (рис. 13). Техника определения. Образец, взвешенный на воздухе с точностью до 0,001 г, прикрепляют тонкой проволочкой к коромыслу гидростатических весов и погружают в стакан с дистиллированной водой при температуре 20±1°С. При погружении образца в воду следят за тем, чтобы на его поверхности и на проволоке не было пузырьков воздуха, а сам образец не касался стенок и дна стакана. Стакан ставят на специальную подставку, которая не должна касаться чашки весов. Опущенный в воду образец взвешивают с точностью до 0,001 г. Затем взвешивают проволоку без образца при том же уровне погружения. Плотность вычисляют по формуле
где т — масса образца на воздухе; т1— масса образца с проволокой в воде; m2 — масса проволоки, погруженной в воду; р0 — плотность воды при 20°С.
ВЯЗКОСТЬ
Вязкостью, или внутренним трением, называют свойство жидкости сопротивляться взаимному перемещению ее частиц, вызванному действием приложенной к жидкости силы. Различают вязкость динамическую и кинематическую. Единица динамической вязкости в системе СИ — ньютон-секунда на квадратный метр — равна динамической вязкости такой жидкости, в которой при изменении скорости движения жидкости на 1 м/с на расстоянии 1 м касательное напряжение равно силе в 1 ньютон на квадратный метр (Н-с/м2). Кинематической вязкостью v(м2/с) называют отношение динамической вязкости Т1< к плотности при той же температуре pt
Единица кинематической вязкости в системе СИ — м2/с. Она равна кинематической вязкости такой жидкости, динамическая вязкость которой 1 Н· с/м2, а плотность 1 кг м3. Относительная вязкость есть отношение вязкости исследуемой жидкости к вязкости другой жидкости, принятой за единицу (при данной температуре). Эта величина удобна для сравнения и не является физической характеристикой продукта. Для жидкостей при данной температуре и давлении вязкость— постоянная величина, поэтому ее значение включают в стандарты для многих продуктов производства химической промышленности. С повышением температуры вязкость уменьшается, с понижением возрастает. В связи с этим измерять ее следует при температуре, указанной в стандартах или технических условиях на материал (обычно при 20°С). Вязкость растворов полимеров зависит от их концентрации и молекулярной массы: при одинаковой концентрации растворов вязкость повышается с увеличением молекулярной массы полимера. Определение вязкости растворов полимеров предусмотрено пофазным контролем многих технологических процессов. Определение вязкости разбавленных растворов полимеров проводят в соответствии ГОСТ 18249—72. Приборы для определения вязкости, называют вискозиметрами. Принцип их действия основан на истечении столба исследуемой жидкости под действием силы тяжести. Наибольшее распространение в техническом анализе получили капиллярные вискозиметры, например, марки ВПЖ-1, ВПЖ-2, ВПЖ-4. Капиллярный вискозиметр ВПЖ-4 (рис. 14) представляет собой стеклянную 1_Г-эбразную трубку, в колено 2 которой впаян капилляр 7, соединенный с шарообразным резервуаром 4. Контрольные метки 5 и 6 служат для наблюдения времени истечения жидкости. Колено 1 и резервуар 8 служат для наполнения вискозиметра испытуемой жидкостью.
Динамическую вязкость вычисляют по формуле
где ∆p— разность давлений на концах капилляра; r — радиус капилляра; l — длина капилляра; V— объем жидкости, протекающей через капилляр; Ʈ — время истечения жидкости. Если жидкость вытекает из капилляра под действием собственной массы, то ∆p=gНр, гдеg— ускорение силы тяжести; Н — разность уровней жидкости в коленах прибора. Тогда
где р — относительная плотность жидкости. Величины V, lи r, а также высота столба жидкости постоянны для каждого вискозиметра. Отсюда
ВеличинаК— постоянная вискозиметра, ее значение указывают в паспорте, приложенном к вискозиметру. Динамическую вязкость ƞ(Н·с/м2) разбавленных растворов полимеров рассчитывают по формуле
где K — постоянная вискозиметра; Ʈ — среднее время истечения раствора или растворителя; р — плотность раствора полимераили растворителя при температуре испытания, определяемая пикнометрическим методом. Кинематическую вязкость раствора полимера v(м2/с) рассчитывают по формуле V = КƮ Техника определения. Перед работой вискозиметр тщательно моют и сушат в сушильном шкафу. Для работы прибор укрепляют вертикально в термостате, в котором находится вода с требуемой по стандарту температурой. Для измерения температуры термостата применяют термометр с ценой деления 0,1°С. Исследуемый раствор полимера (~10 мл) вводят пипеткой в широкое колено вискозиметра 1 (см. рис. 14). После наполнения выдерживают вискозиметр в термостате в течение 15 мин. Затем через резиновую трубку, надетую на колено 2, засасывают раствор полимера в шарик 3. После этого начинают спуск жидкости. Когда уровень жидкости в колене 2 пройдет мимо верхней метки 5, пускают в ход секундомер и следят за опусканием жидкости в колене от верхней до нижней метки 6. Секундомер останавливают в тот момент, когда жидкость пройдет нижнюю метку, и замечают время истечения жидкости от верхней до нижней метки. Измерения повторяют не менее 3 раз (время истечения не должно отличаться более чем на 0,4 с) и находят среднее арифметическое. Если нужно получить относительную вязкость х, определяют динамическую вязкость растворителяƞ0 и раствораƞпри одинаковых условиях (температура термостата, объем, взятый на испытание) и рассчитывают по формуле
4.3. ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
Температурой плавления называют температуру, при которой вещество из твердого состояния переходит в жидкое. Чистое вещество имеет строго определенную температуру плавления. Поэтому температура плавления характеризует степень чистоты продукта. На практике определяют температурный интервал плавления, т. е. интервал между началом плавления — появлением первой капли жидкости и концом плавления, когда все вещество превращается в жидкое состояние. Для чистого продукта температурный интервал составляет 1—3°. Примеси посторонних веществ и присутствие влаги изменяют температуру плавления, расширяют температурный интервал. Поэтому перед определением температуры плавления вещество предварительно перекристаллизовывают и высушивают, затем измельчают в тонкий порошок, так как мелкие частицы плавятся быстрее. Техника определения (по ГОСТ 18995.4—73). В сухую капиллярную трубочку длиной 40—60 мм и внутренним диаметром ~1 мм, запаянную с одного конца, помещают измельченное сухое вещество. Для этого зачерпывают его открытым концом капилляра, погруженным в вещество, и уплотняют многократным опусканием капилляра через стеклянную трубку диаметром 60—80 мм и длиной 1 м, поставленную вертикально на стеклянную пластинку. Уплотнение повторяют до получения слоя вещества высотой 2—3 мм.
Капилляр 4 с веществом помещают в прибор, изображенный на рис. 15. В круглодонную колбу 1 с длинным широким горлом диаметром 30—40 мм и длиной 200 мм помещают широкую пробирку 2 и лабораторный термометр 3, вставленный в пробирку через пробку. Пробирку укрепляют с помощью пробки в колбе так, чтобы она отстояла от дна на 1 см. Колбу на 2/3 объема ее шара заполняют глицерином. Прибор с лабораторным термометром нагревают над асбестовой сеткой на газовой горелке до температуры, на 20—30° ниже предполагаемой температуры плавления. Нагревание регулируют так, чтобы температура поднималась на 1—2°С в 1 мин. Когда температура будет на 10° ниже ожидаемого нижнего предела температурного интервала плавления, лабораторный термометр быстро заменяют на термометр с ценой деления 0,5° с прикрепленным к нему заполненным капилляром. Капилляр прикрепляют резиновым колечком так, чтобы находящееся в капилляре вещество было расположено на уровне середины шарика ртути термометра. За 5° до ожидаемого нижнего интервала плавления нагревание несколько ослабляют и в это время внимательно наблюдают за состоянием вещества. Появление первой капли жидкости и образование мениска считают началом плавления. Конец плавления отмечают, когда исчезнут последние крупинки твердого вещества.
4.4. ТЕМПЕРАТУРА КРИСТАЛЛИЗАЦИИ
Температурой кристаллизации называют наиболее высокую температуру перехода вещества из жидкого состояния в твердое. Ее определяют, охлаждая расплавленное вещество. Химически чистое вещество имеет определенную температуру кристаллизации, а примеси ее понижают. Поэтому температура кристаллизации может служить для оценки чистоты веществ, имеющих низкую температуру плавления или находящихся при комнатной температуре в жидком состоянии. Техника определения. Для определения температуры кристаллизации (ГОСТ 18995.5—73) в пределах 30—150°С применяют прибор Жукова (рис. 16), представляющий собой стеклянный плоскодонный сосуд с двойными стенками, между которыми создан вакуум, чем обусловлено замедленное охлаждение содержимого прибора.
Анализируемый продукт, расплавленный и нагретый на 10—20°С выше предполагаемой температуры кристаллизации, наливают на 3/4 высоты в подогретый прибор Жукова. В отверстие прибора вставляют термометр с ценой деления 0,1° укрепленный на плотно пригнанной пробке так, чтобы ртутный шарик находился посередине слоя испытуемого продукта. Прибор с расплавленным продуктом оставляют медленно остывать. Когда его температура будет на 3—4°С превышать предполагаемую температуру затвердевания продукта, содержимое прибора интенсивно встряхивают, чем вызывают быструю кристаллизацию вещества. В это время температура резко возрастает до максимального значения и остается неизменной в течение некоторого времени, а затем под влиянием внешнего охлаждения начинает понижаться. Температурой кристаллизации считают максимальную температуру, которая остается неизменной в течение короткого времени. Допустимое расхождение между двумя определениями для одного и того же образца не более 0,2°. 4.5. ТЕМПЕРАТУРА КАПЛЕПАДЕНИЯ
В отличие от кристаллических тел аморфные полимеры и смолы плавятся в некотором интервале температур. С повышением температуры твердые смолы размягчаются, и установить точную границу перехода из нетекучего состояния в текучее трудно. Поэтому в большинстве применяемых методов за температуру плавления смол принимают температуру каплепадения, при которой капля полимера отделяется от равномерно нагретой массы испытуемого вещества под действием собственного веса. Температуру каплепадения определяют в приборе Уббеллоде (рис. 17). Он состоит из стеклянного термостата 3, заполненного этиленгликолем и снабженного обратным холодильником 2, термометра 1 с прикрепленным металлическим патроном
4 и металлической чашечки 5 с отверстием в нижней части. Техника определения. Во внутреннюю часть термостата наливают этиленгликоль, уровень которого должен быть ниже дна внутренней пробирки на 10—15 мм. Термостат с обратным холодильником укрепляют на конусномэлектроподогревателе. Включают прибор и нагревают жидкость в термостате при кипении не менее 15 мин. Испытуемую пробу смолы грубо измельчают и загружают в металлическую чашечку на 3/4 ее объема. Чашечку берут щипцами и осторожно, избегая перегрева или горения смолы, нагревают. Смоле необходимо придать лишь пластичность, чтобы можно было термометр погрузить в смолу, прикрепив чашку к верхней части патрона. Смолу, выступающую из отверстия в нижней части чашки, отрезают горячим ножом. Наполненную смолой чашку с термометром укрепляют в термостате. Смола быстро нагревается и, незадолго до падения капли, показывается из нижнего отверстия чашки. Температуру, при которой капля расплавленной смолы проходит через отверстие в дне чашки и падает, считают температурой каплепадения. За результат принимают среднее арифметическое трех определений. Расхождения между определениями не должны превышать. При разборке прибора чашку немного подогревают и осторожно снимают с патрона термометра. Извлекают незатвердевшую смолу, отверстие прочищают латунным шилом, а чашку помещают в спирт или ацетон для полной очистки.
4.6. ТЕМПЕРАТУРА РАЗМЯГЧЕНИЯ СМОЛ
В некоторых случаях для синтетических смол, не имеющих резко выраженного перехода из твердого состояния в жидкое, определяют температуру размягчения (условное понятие). Наиболее распространены для определения температуры размягчения смол методы Кремера — Сарнова и «кольца и шара». Определение температуры размягчения по Кремеру — Сарнову. Слой смолы высотой в 5 мм, находящийся под давлением 5 г ртути, нагревают в стеклянной трубке и отмечают температуру, при которой ртуть прорывается через размягчившуюся смолу.
Прибор Кремера—Сарнова (рис. 18) состоит из двух стеклянных стаканов: наружного / диаметром 8 см и высотой 15 см и внутреннего 2 диаметром 6 см и высотой 10 см. Внутренний стакан укрепляется при помощи специального диска 4 и имеет крышку с пятью отверстиями. В одно отверстие вставляют термометр 3, в остальные — стеклянные трубки 5 внутренним диаметром 6 мм и высотой 5 мм, укрепленные при помощи корковых пробок. В наружный стакан наливают слой глицерина высотой 5 см. Термометр укрепляют так, чтобы шарик ртути находился на уровне испытуемой смолы. Техника определения. Стеклянные трубки ставят на стеклянную пластинку, смоченную водой. В трубки наливают расплавленную смолу так, чтобы сверху образовался небольшой выступ, который после застывания срезают нагретым ножом. Наполненные смолой стеклянные трубки при помощи резиновой трубочки соединяют встык со стеклянными трубками такого же диаметра, но высотой 10 см. В большие трубки наливают по 5 г ртути и вставляют в аппарат так, чтобы смола в трубках находилась на одном уровне с ртутным шариком термометра. После этого аппарат ставят на треножник и постепенно нагревают на асбестовой сетке, повышая температуру на 1—2° в 1 мин, до тех пор, пока ртуть под давлением собственной тяжести не прорвется через слой размягченной смолы на дно стакана. В этот момент отмечают показания термометра. Температуру, при которой капля ртути падает на дно, считают температурой размягчения. Расхождение между двумя определениями не должно превышать 1 °С.
4.7. ТЕМПЕРАТУРА КИПЕНИЯ
Температурой кипения называют температуру, при которой упругость паров вещества становится равной атмосферному давлению, т. е. жидкость кипит при нормальном давлении 100 кПа (760 ммрт.ст.). Химически чистое вещество имеет определенную температуру кипения. Поэтому по температуре кипения судят о качественном и количественном составе вещества.
Рис. 19. Прибор для определения температуры кипения методом перегонки
Температуру кипения определяют микрометодом и методом перегонки, когда за начальную температуру кипения принимают температуру, при которой в приемник упала первая капля жидкости, за конечную— температуру, при которой в приемник перешло 95% жидкости. Работа 1. Определение температурных пределов перегонки (ГОСТ 18995.7—73). Определение проводят в приборе (рис. 19),-состоящем из круглодонной колбы 1 емкостью 100 мл с отводной трубкой, отходящей от середины горла под углом 75° к его нижней части, термометра 2, приемника 4, в качестве которого служит цилиндр емкостью 100 мл с делением в 1 мл, холодильника 3. Для измерения температуры рекомендуется применять укороченный термометр с ценой деления 0,5°. Техника определения. В колбу для перегонки с помощью цилиндра наливают 100 мл исследуемой жидкости так, чтобы жидкость не попадала на стенки колбы и особенно в отводную трубку. Во избежание перегревания жидкости и задержки кипения в перегонную колбу опускают несколько запаянных с одного конца капилляров. В колбу вставляют термометр, укрепленный в пробке таким образом, чтобы ртутный шарик находился на уровне отводного отверстия трубки. Колбу укрепляют на штативе, к отводной трубке присоединяют холодильник, соединенный с чистым и сухим приемником. Подготовленный таким образом прибор нагревают на бане, температура которой должна быть примерно на 20° выше ожидаемой температуры кипения вещества. Прибор с исследуемым веществом нагревают постепенно и ведут наблюдения за температурой и жидкостью. В момент начала кипения температура жидкости быстро поднимается и останавливается на некоторой определенной величине. Температуру, которую показывает термометр при падении в приемник первой капли жидкости, отмечают как начало кипения. Дальнейшее нагревание ведут так, чтобы отгонялось 3—4 мл жидкости в 1 мин. При перегонке необходимо все время наблюдать за показаниями термометра. Для чистых веществ температура перегонки 95% жидкости должна оставаться постоянной. Перегонку заканчивают, когда в приемник перегонится 95 мл исследуемой жидкости, и отмечают конечную температуру кипения. Работа 2. Определение температуры кипения микрометодом.Техника определения. В прибор помещают (рис. 15) тонкостенную стеклянную трубочку диаметром 3 мм и длиной ~80 мм, запаянную с одного конца, и прикрепляют ее резинкой к термометру. В трубочку наливают несколько капель испытуемой жидкости и вставляют капилляр диаметром 1 мм, открытый снизу и имеющий перетяжку. Назначение капилляра— предохранить жидкость от перегревания, чему способствует небольшое количество воздуха, находящегося в нижней части капилляра. Термометр с трубочкой помещают в прибор и нагревают. Незадолго до температуры кипения из капилляра начинают выделяться отдельные пузырьки воздуха, число которых быстро увеличивается; когда жидкость нагреется до температуры кипения, появляется непрерывная цепочка маленьких пузырьков пара анализируемого вещества. В этот момент определяют показание термометра, которое соответствует температуре кипения жидкости.
4.8. ТЕМПЕРАТУРА ВСПЫШКИ И ВОСПЛАМЕНЕНИЯ
Температурой вспышки называют температуру, при которой пары вещества, нагреваемого в определенных условиях, образуют с окружающим воздухом смесь, вспыхивающую при соприкосновении с пламенем. Если вещество нагревать выше температуры вспышки, то наступает момент, когда при поднесении пламени оно загорается. Температура, при которой вещество загорается и горит не менее 5 с, называюттемпературой воспламенения. Температура вспышки и температура воспламенения характеризуют степень огнеопасности вещества, содержание в нем легко испаряющихся веществ. Для определения температуры вспышки применяют приборы открытого и закрытого типов. Температура вспышки одного и того же вещества в аппаратах открытого типа всегда несколько выше, чем в аппаратах закрытого типа. Это объясняется тем, что в последних испарение вещества происходит в сосуде и давление паров, необходимое для создания воспламеняющейся смеси с воздухом от поднесения пламени, достигается значительно раньше, чем в приборах открытого типа, в которых образующиеся пары свободно диффундируют в воздухе. В ГОСТе обычно указан прибор, на котором следует определять температуру вспышки. Температура воспламенения определяется только в приборах открытого типа, где доступ воздуха к поверхности достаточен, чтобы обеспечить горение. Наиболее распространенным аппаратом открытого типа является прибор Бренкена, а закрытого — аппарат Мартенса — Пенского.
Работа 1. Определение температуры вспышки и температуры воспламенения в аппаратах открытого типа (ГОСТ13921—68). Прибор Бренкена (рис. 20) состоит из железного тигля 1 диаметром 63—65 мм, песчаной бани 3, термометра 2 длиной 300 мм, градуированного от 0 до 360°С с ценой деления 1°; зажигательной трубки 4, к которой подводится газ. Прибор имеет шаблон для приливания определенного количества жидкости. Техника определения. Тигель вымывают, сушат и устанавливают в песочную баню так, чтобы между его дном и дном бани оставался слой песка толщиной 5—8 мм, а верхний уровень песка на 10 мм не доходил до верхнего края тигля. Термометр укрепляют в лапке штатива в строго вертикальном положении. В тигель заливают исследуемое вещество, охлажденное до комнатной температуры. Прибор окружают кожухом и помещают в такое место, где нет заметного движения воздуха и свет несколько затемнен, чтобы вспышка была хорошо видна. Баню медленно подогревают. Когда температура бани будет на 10° ниже ожидаемой температуры вспышки, пламенем зажигательной трубки через каждые 2° обводят по краям тигля, делая два оборота по ходу и против часовой стрелки.
Длина пламени для зажигания должна быть 3—4 мм, а длительность каждого испытания не более 2—3 с. За момент вспышки принимают появление над веществом голубоватого, быстро исчезающего пламени, сопровождающегося легким хлопком (взрывом). Температура, соответствующая этому моменту, является температурой вспышки. Далее можно определять температуру воспламенения продукта. Для этого продукт продолжают нагревать и через каждые 2° пламенем зажигательной трубки проводят над поверхностью тигля. Замечают температуру, при которой продукт воспламеняется и горит не менее 5 с,—это температура воспламенения продукта. Работа 2. Определение температуры вспышки в аппаратах закрытого типа.Прибор Мартенса — Пенского (рис- 21) состоит из медного резервуара 1 с плоским дном, внутренним диаметром 50 мм, высотой 55 мм, с кольцевой сеткой на внутренней стороне стенки (до нее наливают испытуемый продукт); крышки резервуара 2, имеющей тубус 8 (для термометра 5); мешалки 4 на гибкой пружинной ручке; зажигательной лампочки 3, которая при повороте рукоятки 6 с механизмом 7 перемещается через отверстие в крышке в свободное от жидкости пространство резервуара; чугунной воздушной бани 10, окруженной металлической рубашкой 9, защищающей ее от потери тепла. Техника определения. Перед определением в тщательно промытый и высушенный резервуар заливают исследуемый продукт до кольцевой метки, помещают в гнездо чугунной воздушной бани, закрывают его чистой, сухой крышкой и вставляют термометр. После этого прибор медленно нагревают. Продукт изредка перемешивают вращением мешалки. При температуре на 10° ниже ожидаемой температуры вспышки через каждые 2° поворачивают . рукоятку 6 и зажигательная лампочка наклоняется в заполненное паром пространство резервуара. Во время испытания перемешивание прекращают. Моментом вспышки считают появление синего пламени над всей поверхностью продукта. После получения первой вспышки испытание продолжают, повторяя зажигание в тех же условиях через 2°. Если при этом вспышки не произойдет, то испытание повторяют заново. За температуру вспышки принимают показания термометра в момент первого появления синего пламени над поверхностью исследуемого вещества при двух параллельных определениях. В том случае, если испытанию подвергают неизвестный продукт, делают предварительное определение температуры вспышки, после чего проводят повторное определение, как описано выше.
ГЛАВА 5
МЕТОДЫ ОПРЕДЕЛЕНИЯ ВЛАГИ
Содержание влаги часто определяют в техническом анализе сырья, вспомогательных материалов, готовой продукции. Такое определение необходимо для правильного расчета содержания основного компонента анализируемого продукта. Выбор метода определения зависит от свойств анализируемого вещества. В веществах, стойких к повышенной температуре, влагу определяют высушиванием до постоянной массы. Метод Фишера позволяет быстро и точно определять любые количества воды в различных органических и неорганических соединениях. Метод Дина и Старка позволяет определять влагу в смолах и пресс-порошках, а также во многих органических соединениях. Этим методом пользуются при анализе веществ, содержащих более 10% воды.
|
||
|
|
Последнее изменение этой страницы: 2018-05-10; просмотров: 1662. stydopedya.ru не претендует на авторское право материалов, которые вылажены, но предоставляет бесплатный доступ к ним. В случае нарушения авторского права или персональных данных напишите сюда... |