|
Студопедия КАТЕГОРИИ: АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция |
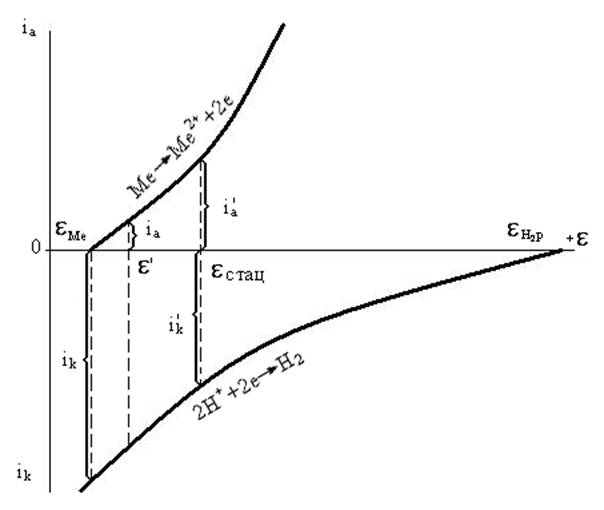
Кинетика электродных процессовВ общем случае скорость коррозии металлов определяется скоростями протекания сопряженных электрохимических реакций при стационарном потенциале: iкорр = iк = iа При записи уравнений парциальных токов необходимо учитывать детальный механизм катодных и анодных реакций в данных условиях. В ряде случаев уравнение скорости коррозии можно свести к виду: iкорр = iк2 = iа1 где iк2 - скорость восстановления окислителя (деполяризатора); iа1 - скорость растворения (ионизации) металла. Исходя из известного из химической кинетики уравнения для скорости химической реакции: v = k C exp [-W/RT], где k - константа скорости реакции, С - концентрация взаимодействующих частиц, W - энергия активации, и с учетом того, что для электрохимической реакции энергия активации зависит от электродного потенциала: Wа = Wао - *а1nF и Wк = Wко + *к2nF, где Wао и Wко - часть энергии активации, не зависящая от потенциала, *а1 и *к2 - кажущиеся коэффициенты переноса, учитывающие долю влияния потенциала на анодную и катодную реакции, для коррозионных процессов, скорость обеих сопряженных реакций которых лимитируется стадией переноса электрона, можно получить: iкорр = Kк2 aOxexp( -*к2Fстац/RT) = Kа1exp( *а1Fстац/RT), где aOx - активность окислителя в коррозионной среде,Kк2 и Kа1 - коэффициенты. Например, при коррозии цинка в кислых растворах, когда скорость выделения водорода на цинке лимитируется стадией переноса электрона и *к2 = к2 = 0,5, а скорость растворения цинка - стадией отщепления второго электрона, т.е. *а1 = 1 +а1 = 1,5 (при а1=к2= = 0,5), тогда уравнение скорости коррозии можно записать в виде:  iкорр = Kк2 aH+exp( -0,5Fстац / RT) = Kа1exp( 1,5Fстац / RT) Из него следует уравнение для стационарного потенциала: стац = (2,3 R T/2F )lg (Kк2 / Kа1)+ (2,3 R T/2F) lg aH+ = const - 0,029 pH. Таким образом, в рассматриваемом случае в отличие от равновесного потенциала металлического электрода, стационарный потенциал не зависит от активности ионов металла, но зависит от pH раствора. Это уравнение хорошо подтверждается экспериментальными данными Ли Ун Сока. Подставляя уравнение для стац в уравнение для плотности тока коррозии, получаем зависимость последней от кислотности раствора: iкорр = K aH+0,75 При коррозии железа в слабокислых растворах скорость восстановления окислителя лимитируется стадией диффузии, т.е. доставкой ионов Н+ к поверхности металла. Следовательно, в данном случае плотность тока коррозии будет равна плотности предельного тока восстановления ионов водорода и плотности тока ионизации металла: iкорр = iпрH+ = Kа1exp(стац *а1F / RT ) (3.11.) откуда следует: стац =( - RT / *а1F) ln KFe + (RT / *а1F) ln iпрH+, (3.12) где KFe = Kа1. Таким образом, стационарный потенциал и плотность тока коррозии железа в данном случае зависят от предельного тока и, следовательно, от интенсивности перемешивания раствора. Коррозионное поведение амальгамы натрия в кислой среде характеризуется преимущественно анодной реакцией: Na(Hg) - e = Na+ и в меньшей мере катодной реакцией: 2 H+ + 2e = H2 . В соответствии с уравнением для плотности тока коррозии, пренебрегая скоростями разряда ионов натрия и ионизации водорода, можно записать: iкорр = Kа1 aNa(Hg)exp(*а1Fстац/RT) = Kk2 aH+exp(-*к2Fстац/RT) В связи с высокой плотностью тока обмена на амальгаме натрия в водных растворах и высоким перенапряжением выделения водорода значение стационарного потенциала практически не отличается от равновесного потенциала амальгамного электрода: стац p = o ам +( RT/F)ln (aNa+ / aNa(Hg)) Подставляя это уравнение в предыдущие, получим выражение для скорости разложения амальгамы: iкорр Kk aH+ (aNa+/аNa(Hg))-*к2, (3.13.) Из уравнения следует, что скорость коррозии в кислых растворах зависит от pH. Учитывая, что *к2 = 0,5, получим при постоянстве aH+ и aNa+ уравнение iкорр K aNa(Hg) (3.14.) которое для амальгам щелочных металлов в кислых растворах экспериментально подтвердили Бренстед и Кейн. В щелочных растворах при pH > 10, когда выделение водорода происходит в результате разряда молекул воды, скорость коррозии амальгамы не зависит от pH раствора. При этом она линейно зависит от концентрации. Для объяснения этого факта предложен химический механизм разложения амальгам в щелочных растворах. Предполагается непосредственное взаимодействие амальгамы с водой по реакции: Na(Hg) + H2O = NaOH + 1/2 H2 Скорость разложения амальгамы в соответствии с этой реакцией: iкорр = aNa(Hg) aH2O не зависит от состава и pH раствора. Химический механизм также доказан и для процессов растворения амальгам щелочных и щелочноземельных металлов в щелочах; железа, марганца, хрома и хромистых сталей в кислотах. При химическом механизме скорость процесса не зависит от потенциала и не наблюдается соответствия между количеством прошедшего электричества и количеством растворившегося металла (Я.М. Колотыркин, Т.Р. Агладзе, В.Н. Коршунов). В общем случае скорость коррозии можно представить уравнением: iкорр =iэх + iхим , где iэх - скорости взаимодействия по электрохимическому механизму; iхим -скорости взаимодействия химическому механизму. Коррозионные диаграммы Метод графического изображения самопроизвольного окисления металлов при помощи поляризационных кривых разработан А.Н.Фрумкиным, А. И. Шултиным, Я.М. Колотыркиным и их учениками. Один из очень характерных случаев - окисление металла катионами водорода (точнее гидроксония H3O+) представлен на рис. 3.3. Анодная поляризационная кривая выражает скорость окисления металла Me Me2+ в зависимости от сдвига потенциала в положительную сторону от равновесного потенциала равнMe его в данном растворе. Катодная кривая К выражает зависимость между сдвигом потенциала в отрицательную сторону от равновесного потенциала выделения водорода равнH2 и скоростью восстановления 2H+ + 2e H2. Как видно из рис. 3.3., потенциал металла, подвергающегося действию окислителя, не может стать равновесным. Невозможность совместить на одном электроде два равновесия, требующих двух различных потенциалов, приводит к установлению стационарного компромиссного потенциала.
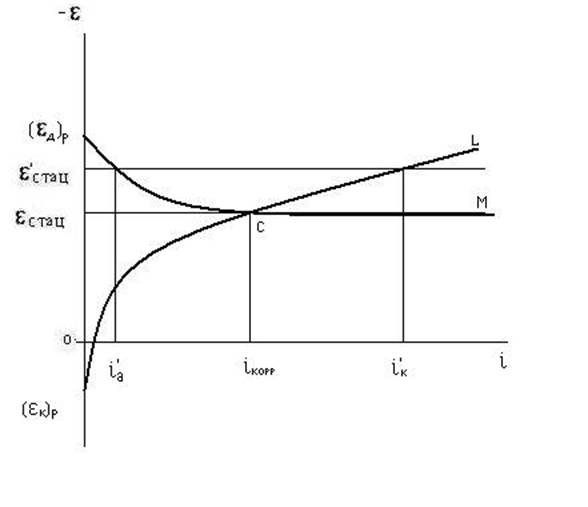
Поляризационные кривые (рис. 3.4.) позволяют легко найти стационарный потенциал, удовлетворяющий равенству скоростей окисления и восстановления, т.е. равенству плотностей анодного и катодного токов на поверхности электрода. Если потенциал электрода примет значение стац, установится стационарное состояние, и окисление металла будет протекать с некоторой постоянной скоростью. Постоянное во времени значение стац реализуется лишь в том случае, когда концентрации ионов металла и окислителя в растворе будут оставаться неизменными.
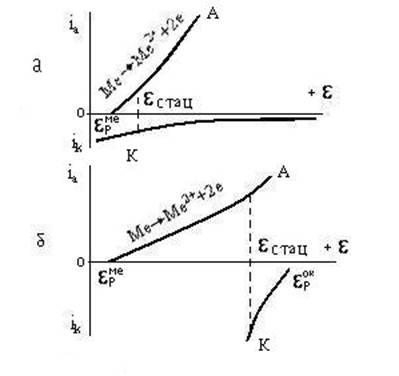
Из рис. 3.3. явствует, что скорость окисления металла должна зависеть от разности между равновесным окислительновосстановительным потенциалом окислителя (в данном случае равнH2) и равновесным потенциалом окисляемого металла равнMe. Чем эта разность больше, тем при данном наклоне поляризационных кривых (т.е. при данном перенапряжении процессов окисления и восстановления) больше скорость окисления металла. Кроме того, скорость окисления зависит и от наклона поляризационных кривых, т.е. от перенапряжения обоих электродных процессов. Очевидно, что в рассматриваемом случае "плотность тока" на поляризационной диаграмме выражает только удельную скорость окисления металла iа и восстановления окислителя iк. Стационарный потенциал может быть близок к равновесному потенциалу металла или окислителя при большом различии в перенапряжении анодного и катодного процессов. На рис. 3.4.а. приведена диаграмма для случая, когда перенапряжение катодной реакции - восстановление окислителя - велико (кривая К). При этом потенциал стац близок к равнMe. Очевидно, что в этом случае скорость коррозии ограничивается скоростью восстановления окислителя (так называемый катодный контроль). На рис. 3.4.б. приведена диаграмма окисления металла, протекающего со значительным перенапряжением. Восстановление окислителя происходит с малым перенапряжением. В этом случае стационарный потенциал близок к равновесному потенциалу окислителя. Скорость реакции при этом ограничивается скоростью окисления металла (анодный контроль). Зачастую металл подвержен действию нескольких окислителей. Например, при коррозии электроотрицательного металла в разбавленном растворе кислоты, соприкасающемся с воздухом, возможны два процесса восстановления окислителей: 2 H+ + 2e H2 и O2 + 2 H2O + 4e 4 OH- Равновесные потенциалы этих процессов значительно отличаются: равнO2 1,229 В, а равнH2 0,00 В, т.е. кислород является более сильным окислителем, чем ион водорода.
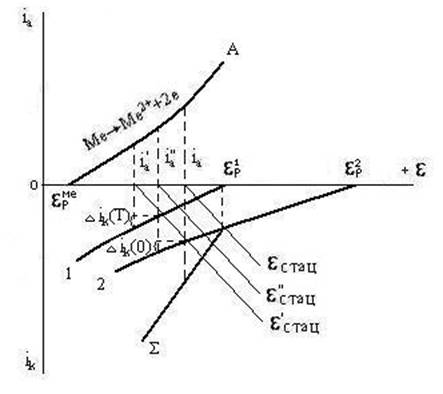
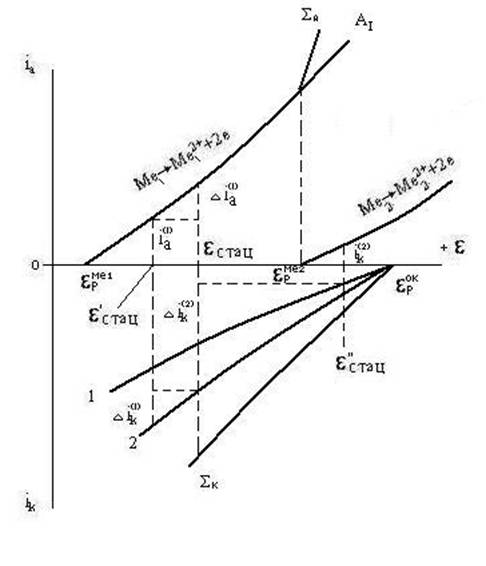
На рис. 3.5. представлен случай окисления металла двумя окислителями (деполяризаторами) разной силы. Катодные кривые восстановления окислителей обозначены 1 и 2; равновесные потенциалы окислителей равн1 и равн2; равновесный потенциал металла равнMe. Если бы в растворе присутствовал только окислитель 1, то установился бы стационарный потенциал 'стац и металл окислялся бы со скоростью, выражаемой плотностью тока iа'. Если бы присутствовал только окислитель 2, то стационарному потенциалу ''стац соответствовала бы скорость окисления iа". В присутствии двух окислителей при потенциалах более отрицательных, чем равн, возможно восстановление обоих окислителей. Поэтому катодные парциальные кривые необходимо суммировать. Кривая представляет сумму скоростей восстановления обоих окислителей при потенциалах, более отрицательных, чем равн1. Учитывая катодную и анодную суммарные поляризационные кривые, найдем стационарный потенциал, отвечающий равенству скоростей окисления и восстановления.
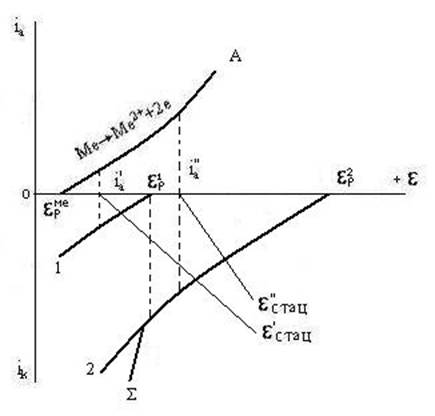
Для данного случая стационарным потенциалом будет ''стац. Это свидетельствует, что металл будет окисляться только окислителем 1. Более сильный окислитель 2 вызывает столь значительное смещение потенциала металла в положительную сторону, что ''стац оказывается положительнее, чем равн1. В этих условиях окислитель 1 не может окислять металл. Примером может служить раствор азотной кислоты, оба иона которой (H+ и NO3-) являются окислителями различной силы. При pH=0 активность катионов водорода равна единице и равн2 = 0. Стандартный окислительно-осстановительный потенциал, отвечающий равновесию NO + O2 NO3-, равен +0,96 В. При таком соотношении потенциалов обоих окислителей коррозия металла, например, железа, происходит только за счет восстановления анионов NO3-. Подобное же соотношение имеет место при окислении многих металлов в нейтральных растворах солей неокислителей, содержащих растворенный кислород воздуха. При pH=7 потенциал равнH2 = -0,41 В, а равнO2 = +0,82 В. Многие металлы в этих условиях окисляются только за счет растворенного в коррозионной среде кислорода. На рис. 3.6. изображена поляризационная диаграмма для совместного действия двух окислителей, равновесные потенциалы которых равн1 и равн2 имеют меньшее различие, чем в выше рассмотренном случае. При данных наклонах анодной кривой А и катодных кривых 1 и 2 каждый из окислителей порознь окислял бы металл с различной скоростью: окислитель 1 - со скоростью i'а при потенциале 'стац, окислитель 2 - со скоростью i''а при потенциале ''стац. Суммируя катодные кривые, получим кривую , относящуюся к совместному восстановлению окислителей 1 и 2. В присутствии обоих окислителей установится стационарный потенциал стац, отвечающий скорости коррозии металла iа равной суммарной скорости восстановления окислителей. Скорость коррозии iа больше парциальной скорости i'а или i''а. Скорость же восстановления каждого из окислителей уменьшается вследствие их совместного действия.
Возможно и такое соотношение равновесных потенциалов металла и окислителей, когда один из окислителей вообще не может окислять металл. Так, например, в растворах кислот-неокислителей медь не может растворяться в отсутствии растворенного кислорода. Очевидно, что окислитель, присутствующий в растворе, сдвигает потенциал металла от равновесного значения в электроположительную сторону. Попытки измерить равновесный потенциал металла, например, цинка или меди в растворе собственных солей в присутствии растворенного кислорода воздуха, обречены на неудачу, т.к. измерение даст величину стационарного (коррозионного) потенциала, а не равновесного. Большой практический интерес представляет окисление двух различных металлов, находящихся в контакте друг с другом. На рис. 3.7. приведена диаграмма окисления двух металлов одним окислителем, где равнMe1 и равнMe2 - равновесные потенциалы металлов, равнок - равновесный потенциал окислителя. При потенциалах более положительных, чем равнMe2, возможно окисление обоих металлов, что отвечает суммарной анодной поляризационной кривой А. Перенапряжение восстановления окислителя на различных металлах неодинаково. Катодная кривая 2 восстановления окислителя на металле Me2 идет более полого, что указывает на большее перенапряжение. Если бы металлы не соприкасались друг с другом, то каждый из них окислялся бы со скоростью, определяемой - двумя кривыми: Me1, приняв стационарный потенциал 1стац, окислялся бы со скоростью i1а; Me2 при стационарном потенциале 2стац окислялся бы со скоростью i2а. Но так как металлы находятся в контакте, то восстановление окислителя возможно на каждом из них, и катодные кривые 1 и 2 должны быть просуммированы (кривая k). Рассматривая совместно суммарные кривые А и k, найдем стационарный потенциал стац, при котором суммарная скорость окисления будет равна суммарной скорости восстановления. В данном случае будет происходить окисление только одного металла Me1. Стационарный потенциал стац более отрицателен, чем равнMe2, что и препятствует окислению Me2. Скорость окисления Me1 в результате контакта с Me2 возрастает на величину ia1. Металл Me2 защищен от коррозии за счет контакта с более электроотрицательным Me1. При контакте двух металлов изменяется и скорость восстановления окислителя на каждом из них. На поверхности металла Me1 скорость восстановления уменьшается на величину iк1, а на поверхности металла Me2 - увеличивается на величину iк2. При окислении металлов находящихся в контакте происходит движение электронов от Me1 к Me2 и движение ионов в растворе: катионов от Me1 к Me2 и анионов в противоположном направлении, что обеспечивает поддержание на каждом электроде стационарного потенциала. Такая пара металлов представляет собой короткозамкнутый гальванический элемент. Если электропроводность электролита достаточно велика и переходное сопротивление в месте контакта двух металлов мало, тогда омическое падение напряжения в цепи элемента пренебрежимо мало. При этих условиях может установиться единый потенциал для обоих металлов стац. Тогда в рассматриваемом случае обеспечивается полная катодная защита от коррозии Ме2. Если омическое сопротивление электролита и контакта сравнительно велико, то оба металла будут иметь разные значения потенциалов. Чем большая разность потенциалов сохранится между металлами, тем менее вероятна полная защита металла Me2, т.е. тем положительнее будет его потенциал. Если он окажется положительнее равновесного равнMe2, то будет происходить окисление Me2, хотя и более медленное, чем в отсутствии контакта с Me1, т.е. неполная защита Me2. Прекращение или замедление окисления металла Me2 может быть вызвано сдвигом его потенциала в отрицательную сторону либо за счет контакта с более электроотрицательным металлом (например, железо защищается при контакте с цинком), либо за счет наложения внешней ЭДС.
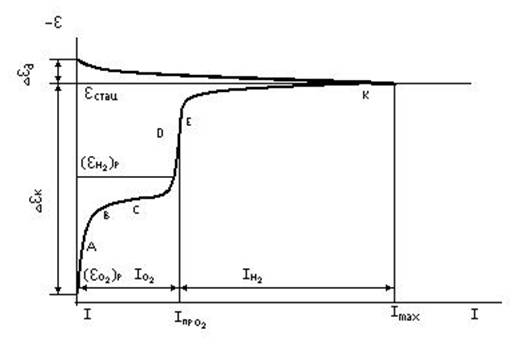
Для графического расчета скорости окисления металла с помощью диаграмм коррозии необходимо знание идеальных поляризационных кривых: а = f(ia) - анодная поляризационная кривая для анодных участков корродирующего металла и к = f(iк) - катодная поляризационная кривая поляризации катодных участков корродирующего металла (рис. 3.8). Если суммарные поверхности катодных и анодных участков равны (Sк= Sa), тогда ia= iк = iкорр. В этом случае точка пересечения поляризационных кривых С соответствует стационарному потенциалу стац и скорости коррозии системы iкорр. Согласно Н.Д. Томашову, скорость коррозии и соотношение катодной и анодной зон можно рассчитать зная стационарный потенциал корродирующего гетерогенного металла и поляризационные идеальные кривые в условиях, близких к условиям коррозии данного металла. Как правило, в реальных условиях наблюдается отличие объемов и площадей анодных и катодных зон. В этом случае определение скорости коррозии и характеристик коррозионного процесса производят с помощью диаграмм коррозии [Ю.Р. Эванс, 1929 г.], построенных в координатах потенциал - сила тока (рис. 3.9). Если Sa Sк, тогда плотности тока на анодных и катодных участках тоже не равны: ia iк. Однако, сила коррозионного тока общая и для катодного, и для анодного процесса: I = ia Sa = iк Sк. Для построения диаграммы Эванса из идеальных поляризационных кривых а = f(ia) и к = f(iк) путем пересчета с учетом площадей анодных и катодных участков находят зависимости а = f(I) и к = f(I). Точка пересечения анодной и катодной кривых C (рис.3.9) отвечает значению максимального коррозионного тока Imax и общему стационарному потенциалу двухэлектродной системы стац при отсутствии омического сопротивления внешней цепи (R 0). Как известно, такие системы называют короткозамкнутыми. ЭДС коррозионного процесса E = кобр - аобр полностью израсходована на преодоление поляризационных сопротивлений анодного и катодного процессов, при этом поверхность металла практически эквипотенциальна. Из диаграммы Эванса легко найти графически значения анодной а и катодной к поляризации. Если омическое сопротивление корродирующей двухэлектродной системы не равно нулю, то поверхность металла не эквипотенциальна. Графическим путем достаточно легко найти значения эффективных потенциалов анодных и катодных участков и омическое падение напряжения при силе тока коррозии. Как известно, процесс электрохимической коррозии состоит из последовательных более простых процессов (стадий): анодного, катодного и процесса протекания электрического тока. Установившаяся скорость процесса, соответствующая силе коррозионного тока I, определяется торможением протекания тока, т.е. сопротивлениями его отдельных стадий, на преодоление которых расходуется начальная разность потенциалов электродных процессов, т.е. ЭДС. Ранее с учетом теории гальванического элемента показано, что сила тока Iкорр = E/(R + r) . В данном случае сопротивление внутренней цепи складывается из удельных поляризуемостей анодного и катодного процессов: r = Pа + Pк. В свою очередь, P = /I при I=1, тогда Iкорр = E/(R + Pа + Pк);
E = Iкорр /(R + Pа + Pк) = Uом +а + к. Поляризуемость складывается из поляризуемости электродной реакции Pр и поляризуемости диффузии Pд, т.е. P = Pр + Pд. Долю сопротивления стадии коррозионного процесса по отношению к общему сопротивлению процесса называют степенью контроля [Н.Д. Томашов, 1947 г.] и обозначают СА , СК и СR - это соответственно степени анодного, катодного и омического контроля: Са = {а/(кобр - аобр)} 100,%; Ск = {к/(кобр - аобр)} 100,% ; СR = {Uом/(кобр - аобр)} 100,%. Контролирующим называют процесс (стадию), кинетика которого определяет скорость коррозии. Для определения контролирующего процесса сравнивают Са, Ск и СR или а, к и Uом. Знание контролирующей стадии электрохимической коррозии позволяет целенаправленно воздействовать на эту контролирующую стадию процесса, тормозя ее и таким образом весь процесс коррозии.
3.2.3. Кинетика катодных процессов Типы катодных реакций при электрохимической коррозии металлов: - восстановление катионов 2 H+ + 2e = H2 , Cu2+ + 2e = Cu , Ag+ + e = Ag , Cu2+ + e = Cu+, Fe3+ + e = Fe2+, Fe2+ + 2e = Fe; - восстановление анионов S2O82-+ 2e = 2SO42-, 2HSO3-+ 2H+ 2e = S2O42-+ 2H2O, NO3-+ 3H+ 2e = HNO2 + H2O, Cr2O72-+ 14H+ 6e = 2Cr23++ 7H2O; - восстановление нейтральных молекул O2 + 2H2O + 4e = 4OH- , H2O2 + 2e = 2OH- , Cl2 + 2e = 2Cl- , Br2 + 2e = 2Br- ; - восстановление нерастворимых пленок CuO + H2O + 2e = Cu + 2OH-, Fe3O4 + H2O + 2e = 3FeO + 2OH- Fe(OH)3 + e = Fe(OH)2 + OH-, Fe(OH)2 + 2e = Fe + 2OH- ; - восстановление органических соединений RO + 4H+ + 4e = RH2 + H2O , R + 2 H+ + 2e = RH2 , где R - радикал или органическая молекула. В большинстве случаев электрохимическая коррозия протекает с кислородной ( восстановление растворенного в воде кислорода ) и (или) водородной (восстановление ионов водорода) деполяризацией. Коррозия металлов с преобладанием водородной деполяризации протекает: - при высокой активности ионов водорода в растворе (например, коррозия железа, цинка и многих других металлов в растворах сильных кислот); - при значительной активности корродирующего металла (весьма электроотрицательный потенциал равнMe) - например, коррозия щелочных и щелочноземельных металлов в воде, нейтральных электролитах и щелочах. Механизм реакции катодного выделения водорода. Эта реакция в кислых растворах представлена следующими последовательными стадиями: 1 - диффузия и миграция гидратированных ионов водорода H+ H2O к катодным участкам поверхности корродирующего металла; 2 - разряд гидратированных ионов водорода; 3 а - растворение и диффузия части Hадс - атомов в металле; 3 б - рекомбинация (молизация) адсорбированных атомов водорода или электрохимическая десорбция: е Hадс. + H+.H2O H2 + H2O ; 4 - образование и отрыв пузырька водорода от поверхности металла; 5 - диффузия и перенос конвекцией водорода от катодных участков в объем раствора, с последующей диффузией в атмосферу. В общем случае указанная последовательность стадий может быть представлена схемой: е H+H2O(раств.)H+H2O(пов.)H(адс.)+H2OH2(пов.) H2(газ). В щелочных растворах, в которых активность ионов водорода (точнее, гидроксония) очень мала, электрохимическая стадия заключается в восстановлении молекул воды, концентрация которой у поверхности велика. Поэтому стадия доставки разряжающихся частиц к поверхности отсутствует и последовательность стадий будет следующей: H2O H(адс.) + OH- H2(пов.) H2(газ.) , и, кроме того, появляется дополнительная к перечисленным выше стадия отвода ионов OH- в объем раствора. В подавляющем большинстве случаев концентрационная поляризация связанная с затруднениями доставки водородных ионов к поверхности катода, отсутствует, что обусловлено аномально высокой подвижностью водородных ионов. Кроме этого, происходит интенсивное перемешивание раствора у катода выделяющимся водородом и дополнительный перенос ионов водорода к катоду миграцией. Иногда наблюдается концентрационная поляризация связанная с замедленностью процесса отвода от поверхности металла молекулярного водорода. В нейтральных растворах или при очень больших скоростях коррозии проявляется ионная концентрационная поляризация. Главной причиной катодной поляризации является замедленность катодной реакции, которая приводит к перенапряжению выделения водорода. Для перенапряжения выделения водорода Н2 в широком диапазоне плотностей тока от 10-5до 106 А/м2 выполняется эмпирическое уравнение (И. Тафель, 1905 г.): H2 = a + b lg iк (3.15.) Постоянная a в уравнении Тафеля, т.е. величина перенапряжения при плотности тока, равной единице (iк = 1 А/см2), зависит от природы металла, состояния его поверхности, состава электролита, температуры и характеризует собой степень необратимости процесса на электроде. Чем больше a, тем больше величина перенапряжения при данной плотности тока, тем больше отклонение от обратимого состояния. Значение константы a обычно лежит в пределах от 0,1 до 1,5 В. Величина b характеризуется значениями от 0,03 до 0,12 и в редких случаях превышает 0,12. Перенапряжение выделения водорода существенно зависит от температуры, pH, состава раствора и присутствия в нем поверхностноактивных веществ (ПАВ). С повышением температуры H2 снижается, меняются также константы уравнения Тафеля: a уменьшается, b увеличивается. При малых отклонениях от равновесного состояния величина перенапряжения прямо пропорциональна плотности тока, проходящей через электрод: H2 = k iк. Особенности коррозии металлов с водородной деполяризацией: - сильная зависимость от pH раствора: H2 в кислых растворах снижается, а в щелочных растет, т.к. с уменьшением pH равновесный потенциал водородного электрода возрастает; - значительная зависимость от природы и концентрации катодных примесей или структурных составляющих сплава: чем ниже H2 на вышеназваных примесях или составляющих и чем выше их содержание, тем больше скорость коррозии; - большая зависимость скорости коррозии от адсорбции на поверхности металла веществ, влияющих на строение двойного электрического слоя и величину H2; - протекание процесса с ускорением, т.к. по мере растворения металла обнажаются новые катодные участки, что облегчает процесс катодного восстановления ионов водорода; - незначительная зависимость скорости коррозии от перемешивания электролита, особенно в растворах кислот; - возможность наводороживания металла и возникновения водородной хрупкости его.
Механизм катодного процесса с кислородной деполяризацией Катодный процесс включает в себя следующие последовательные стадии: - растворение кислорода воздуха в электролите; - перенос растворенного кислорода в объеме электролита за счет естественной конвекции или перемешивания; - конвективный перенос кислорода в части приэлектродного слоя Прандтля; - перенос кислорода в диффузионной части приэлектродного слоя или в пленке продуктов коррозии к катодным участкам поверхности металла; - ионизация кислорода: в нейтральных и щелочных средах O2 + 2 H2O + 4e = 4 OH-(водн.) , в кислых растворах O2 + 4 H+(водн.) + 4e = 2 H2O ; - диффузия и конвективный перенос ионов OH- от поверхности металла в объем электролита. Чаще всего при коррозии металла с кислородной деполяризацией самой медленной стадией катодного процесса является диффузия кислорода. При очень больших скоростях подвода кислорода за счет интенсивного перемешивания или при весьма тонкой пленке электролита на поверхности металла скорость катодного процесса лимитирует ионизация кислорода. Довольно часто наблюдается смешанная поляризация (скорость диффузии соизмерима со скоростью ионизации кислорода). При очень малых плотностях тока перенапряжение ионизации кислорода О2 линейно зависит от плотности тока О2 = k2 iк , где k2 - постоянная, зависящая от материала и состояния поверхности катода, температуры и ряда других факторов. При больших плотностях тока О2, также как и Н, имеет логарифмическую зависимость от плотности тока: О2 = a2 + b2 lgiк , (3.16.) где a2 - постоянная, зависящая от материала и состояния поверхности катода, а также от температуры раствора и др., b2 - постоянная, связанная с механизмом возникновения перенапряжения ионизации кислорода. Н.Д. Томашовым измерено перенапряжение ионизации кислорода на различных металлах. Металлы с высоким О2 : Mg, Al, Zn, Cd, Pb; средним: Co, Cr, Ni, Sn, Fe, Cu; низким: Au и Pt. С повышением температуры О2 уменьшается.
|
|||||||||
|
|
Последнее изменение этой страницы: 2018-05-30; просмотров: 445. stydopedya.ru не претендует на авторское право материалов, которые вылажены, но предоставляет бесплатный доступ к ним. В случае нарушения авторского права или персональных данных напишите сюда... |