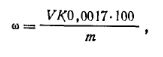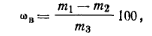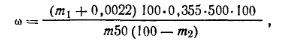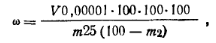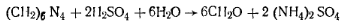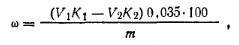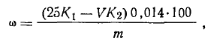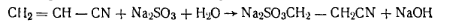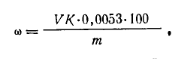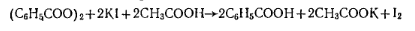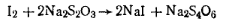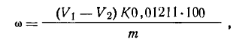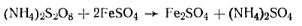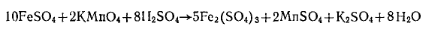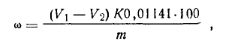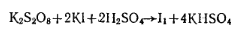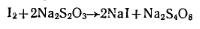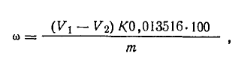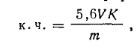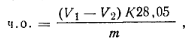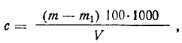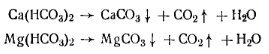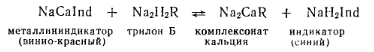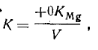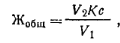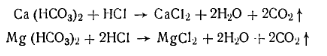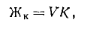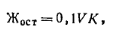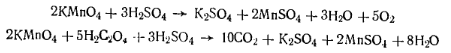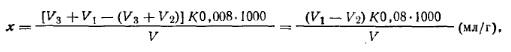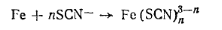|
Студопедия КАТЕГОРИИ: АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция |
Работа 2. Определение содержания свободного аммиака.
Реактивы и оборудование Соляная кислота, 0,1 н. раствор Колба коническая на 500 мл Метиловый оранжевый, 0,1%-ный раствор Ход определения. Навеску 50 г мочевины, взвешенную с точностью до 0,01 г, помещают в коническую колбу емкостью 500 мл, растворяют в 200 мл дистиллированной воды, добавляют 3—5 капель индикатора метилового оражевого и титруют 0,1 н. раствором соляной кислоты до перехода желтой окраски раствора в оранжевую. Массовую долю свободного аммиака ω (%) рассчитывают по формуле
где V— объем 0,1 н. раствора соляной кислоты, израсходованный на титрование; т — масса навески мочевины; К. — коэффициент поправки для 0,1 н. раствора соляной кислоты; 0,0017 — масса аммиака, соответствующая 1 см3 точно 0,1 н. раствора соляной кислоты, г.
Работа3. Определение массовой доли воды.Воду определяют ускоренным методом — облучением мочевины инфракрасными лучами. Оборудование Алюминиевый бюкс Эксикатор Установка с лампой инфракрасного излучения
Ход определения. Навеску мочевины ~5 г взвешивают в алюминиевом бюксе с точностью до 0,0002 г. Бюкс помещают в установку под лампой (см. рис. 22) на расстоянии 15 — 18 см. Сушат при температуре 70 — 75°С в течение 15 мин. Затем бюкс закрывают крышкой, охлаждают в эксикаторе и взвешивают. Массовую долю воды ωв (%) вычисляют по формуле
где т1— масса бюкса с мочевиной до высушивания; т2— масса бюкса с мочевиной после высушивания; т3— масса навески мочевины, взятой на анализ.
Работа 4. Определение массовой доли железа.Массовую долю железа в мочевине определяют колориметрически, методом стандартных серий. Определение заключается в том, что готовят два раствора — испытуемый и эталонный, который имеет точно известную концентрацию определяемого компонента. Fе3+ образует окрашенное комплексное соединение с роданидом аммония, которое хорошо растворимо в изоамиловом спирте, чем и пользуются для того, чтобы его выделить, так как в присутствии ряда посторонних ионов он разрушается.  Сравнением растворов устанавливают массовую долю железа в техническом продукте. Реактивы и оборудование Соляная кислота, р=1,19 Колба мерная иа 100 мл Перманганат калия, 0,1 н. раствор Цилиндр мерный на 50 мл с при- Роданид аммония, 10%-ный раствор шлифованной пробкой Изоамиловый спирт Пипетка на 10 мл Эталонный раствор соли железа
Приготовление эталонного раствора, содержащего Fe2О3, 6,04 г перекристаллизованных железоаммонийиых квасцов помещают в мерную колбу емкостью 1000 мл и растворяют в 200 — 300 мл воды. К раствору прибавляют 25 мл концентрированной горной кислоты и объем жидкости в колбе доводят водой до метки. Раствор тщательно перемешивают, 1 мл полученного раствора содержит 1 мг Fe2О3. Для работы раствор разбавляют в 10 раз, так чтобы 1 мл его содержал 0,1 мг Fe2О3. Ход определения. 10 г испытуемой мочевины, взвешенной с точностью до 0,01 г, растворяют при нагревании в смеси, состоящей из 40 мл дистиллированной воды и 10 мл концентрированной соляной кислоты. К раствору по каплям прибавляют 0,1 и. раствор псрмапганата калия (чтобы окислить Fe+2 в Fе3+) до появления розовой окраски, затем нагревают на водяной бане и кипятят в течение 5 мин. После охлаждения раствор переливают в мерную колбу емкостью 100 мл, доводят объем дистиллированной водой до метки и тщательно перемешивают. В градуированный цилиндр с притертой пробкой емкостью 50 мл вносят пипеткой 10 мл приготовленного раствора, прибавляют 5 Мл 10%-ного раствора роданида аммония или калия, перемешивают, добавляют 10 мл изоамилового спирта и снова перемешивают. Одновременно готовят раствор сравнения. Для этого берут те же объемы воды, соляной кислоты и перманганата калия, кипятят 5 мин, затем переносят в мерную колбу емкостью 100 мл и разбавляют водой до метки. 10 мл приготовленного раствора переносят в цилиндр емкостью 50 мл, прибавляют 0,5 мл эталонного раствора, 5 мл 10%-ного раствора роданида аммония, перемешивают, добавляют 10 мл изоамилового спирта и снова перемешивают. Если окраска раствора, содержащего мочевину, менее интенсивна, чем эталонного раствора, то содержание железа в техническом продукте не превышает нормы, установленной стандартом. Сравнение интенсивности их окраски производят после уравнивания объемов жидкости и взбалтывания содержимого обоих цилиндров. МеламинС3Н6N6(Мг= 126,13) Белое мелкокристаллическое вещество. Температура плавления 354°С. При осторожном нагревании сублимируется, при сильном нагревании разлагается. Растворим в горячей воде; не растворим в этиловом спирте и других органических растворителях. В промышленности меламин получают нагреванием дициапдиамида под давлением в присутствии свободного аммиака. Технические требования по ГОСТ 7579—76: массовая доля меламина в сухом продукте не менее 99,7%; массовая доля воды не более 0,2%; массовая доля железа не более 0,0005%; нерастворимые в воде вещества отсутствуют. Работа 5. Определение массовой доли меламина.Метод основан на взаимодействии меламина с пикриновой кислотой с образованием пикрата меламина. Массовую долю меламина определяют исходя из массы осадка пикрата меламина. Реактивы и оборудование
Ход определения. 1 г растертого в тонкий порошок меламина взвешивают с точностью до 0,0002 г, переносят в стакан емкостью 500 мл и растворяют в 30 мл горячей воды при нагревании почти до кипения в течение 20 мин. Раствор охлаждают и фильтруют через фарфоровую воронку. Фильтр с осадком пятикратно промывают водой порциями по 10 мл, затем фильтрат и промывные воды переводят в мерную колбу емкостью 500 мл, доливают дистиллированной водой до метки и перемешивают. Пипеткой отбирают 50 мл полученного раствора, переносят в стакан емкостью 500 мл с. двумя метками, указывающими объем 200 и 400 мл, и добавляют 5 мл ледяной уксусной кислоты. Раствор разбавляют кипящей водой до метки 200 мл и приливают тонкой струей (без перемешивания) насыщенный раствор пикриновой кислоты до момента образования первых кристаллов пикрата. В этот момент прекращают приливание раствора пикриновой кислоты, стакан с раствором помещают в холодную воду или снег, дают пикрату кристаллизоваться без перемешивания. Через 10 мин приливают раствор пикриновой кислоты до метки 400 мл, жидкость с осадком перемешивают, охлаждают до 20°С и выдерживают при этой температуре 1—1,5 ч. Затем осадок фильтруют через предварительно взвешенный с точностью до 0,0002 г фильтр-тигель с фильтрующей пластинкой № 1 или № 2, отсасывают жидкость и 3—4 раза промывают осадок небольшими порциями насыщенного раствора пикрата меламина. Тигель с осадком сушат в термостате при 105—110°С до постоянной массы. Массовую долю меламина ω (%) вычисляют по формуле где m — навеска меламина; т1— масса осадка пикрата меламина; 0,0022 — растворимость пикрата меламина при температуре 20°С в 400 см3 жидкости, из которой выпадает осадок; 0,355 —множитель для перевода массы пикрата на массу меламина; m2— масса воды.
Работа 6. Определение содержания воды.Воду определяют высушиванием меламина до постоянной массы. Высушивание ведут ускоренным методом — облучением инфракрасными лучами или сушкой в термошкафу при 100—105°С. Методику определения и расчет см. разд. 6.1 или 6.2. Работа 7. Определение массовой доли железа.Массовую долю железа в меламине определяют методом колориметрического титрования, добиваясь одинаковой окраски раствора меламина и воды, к которой постепенно добавляют эталонный раствор железа. Реактивы и оборудование
Приготовление эталонного раствора, содержащего Fe2О3. 0,864 г железоаммоннйных квасцов растворяют в мерной колбе емкостью 1000 мл, подкисляют 25 мл серной кислоты (пл. 1,84) и доводят объем раствора до метки. Берут 10 мл полученного раствора и разбавляют водой до 100 мл. 1 мл этого раствора содержит 0,00001 г железа. Раствор применяют свежеприготовленным. Ход определения. Взвешивают 1 г меламина с точностью до 0,01 г, помещают в коническую колбу емкостью 250 мл и растворяют при кипячении в смеси, состоящей из 80 мл дистиллированной воды, 2 мл бромной воды, содержащей 16 г брома в 1 л, и 5 мл соляной кислоты. Смесь кипятят до удаления запаха брома, охлаждают и переводят в мерную колбу емкостью 100 мл. Доводят объем раствора до метки и перемешивают. 25 мл полученного раствора переносят в градуированный цилиндр с притертой пробкой емкостью 50 мл. Берут другой такой же цилиндр и вносят в него 25 мл раствора, приготовленного разбавлением дистиллированной водой 5 мл концентрированной соляной кислоты до объема 100 мл. Затем в оба цилиндра добавляют по 5 мл изоамилового спирта и по 5 мл 10%-ного раствора роданида аммония. Смесь встряхивают, оба цилиндра ставят на белую бумагу. Если окраска цилиндра, где находится только раствор соляной кислоты, окажется слабее, чем в определяемом растворе, то в первый цилиндр прибавляют из микробюретки эталонный раствор железа, взбалтывают, сравнивают окраску. Так повторяют до тех пор, пока она в обоих цилиндрах не сравняется. Массовую долю железа ω (%) вычисляют по формуле
где V— объем эталонного раствора железа, израсходованный на выравнивание окраски; т — масса навески меламина; 0,00001—масса железа в 1 см3 эталонного раствора; m2 — массовая доля воды. Гексаметилентетрамин(уротропин) (СН2)6N4(Мr= 140,19)
Крупнокристаллический неслеживающийся порошок белого цвета. Улетучивается при нагревании, не плавясь. Растворяется в воде и этиловом спирте. При попадании на кожу вызывает раздражение. В промышленности Гексаметилентетрамин получают конденсацией аммиака с формальдегидом. Технические требования по ГОСТ 1381—73: массовая доля аминов не менее 99,5%; воды не более 0,5%. Работа 8. Определение массовой доли гексаметилентетрамина.Определение основано на реакции кислотного гидролиза. При действии серной кислоты Гексаметилентетрамин разлагается с выделением формальдегида и образованием сульфата аммония:
По массе кислоты, затраченной на образование аммонийной соли, рассчитывают массовую долю гексаметилентетрамина. Молярная масса эквивалента гексаметилентетрамина равна 1/4 массы его моля.
Реактивы и оборудование Серная кислота, 1 н. раствор Бюкс для взятия иавески Гидроксид натрия, 1 н. раствор Колба коническая на 250 мл Метиловый оранжевый, 0,1%-ный Пипетка на 50 мл раствор
Ход определения. Навеску гексаметилена ~ 1 г, взятую с точностью до 0,0002 г, вносят в коническую колбу емкостью 250 мл и приливают 500 мл 1 н. серной кислоты. Нагревают на кипящей водяной бане до исчезновения запаха формальдегида, выделяющегося в результате гидролиза. Реакция продолжается 2—3 ч. Охладив раствор, избыток кислоты оттитровывают 1 н. раствором гидроксида натрия в присутствии индикатора метилового оранжевого. Массовую долю гексаметилентетрамина ω (%) вычисляют по формуле
где v1— объем 1 н. раствора серной кислоты; К1— коэффициент поправки для 1 н. раствора серной кислоты; V2— объем1 н. раствора гидроксида натрия; K2 — коэффициент поправки для 1 н. раствора гидроксида натрия; 0,0350 — масса гексаметилентетрамина, соответствующая 1 см3 точно 1 н. раствора серной кислоты, г; т — навеска гексаметилентетрамина. Акрилонитрил(нитрил акриловой кислоты) СН2=СН—СN (Мr=53,06) Бесцветная подвижная жидкость со слабым запахом. Температура кипения 77,3°С, температура плавления —82°С. По ГОСТ 11097—86 р420 0,8000÷0,8060 г/см3, показатель преломления np20 1,3910÷1,392. Токсичен. Растворяется во многих органических растворителях, в воде практически не растворим. Хранится в присутствии ингибиторов (гидрохинон, пирогаллол). В промышленности акрилонитрил получают окислительным аммонолизом пропилена или прямым синтезом из ацетилена и цианида водорода. Применяют для изготовления синтетического волокна «нитрон» и для сополимеризации с другими мономерами. Для определения содержания акрилонитрила в исследуемом продукте пользуются либо методом Кьельдаля, либо потенциометрическим методом. Работа 9. Определение массовой доли азота по Кьельдалю.Сущность метода состоит в том, что азот органического вещества при нагревании с концентрированной серной кислотой образует сульфат аммония. При действии концентрированного раствора щелочи из сульфата аммония выделяется аммиак, который поглощается определенным количеством титрованного раствора кислоты, а затем избыток кислоты титруют раствором щелочи. Реактивы и оборудование Серная кислота, р=1,84 Метиловый оранжевый, 0,1%-ный Серная кислота, 1 и. раствор раствор Гидроксид натрия, 40%-Нын раствор Бюкс для взятия навески Гидроксид Натрия, 1 н. раствор Колба Кьельдаля Прибор Кьельдаля
Ход определения. Навеску акрилонитрила ~1 г, взятую с точностью до 0,0002 г, помещают в колбу Кьельдаля (см. рис. 45, а), прибавляют кристалл сульфата меди (для ускорения разложения) и вливают 25 мл серной кислоты. Колбу закрывают крышкой, закрепляют с помощью штатива 'в слегка наклонном положении над асбестовой сеткой под тягой и нагревают сначала маленьким пламенем в течение 15 мин, а затем более интенсивно, пока жидкость не начнет кипеть. Нагревание продолжают до тех пор, пока раствор в колбе не станет прозрачным и бесцветным. После охлаждения раствор переносят в перегонную колбу емкостью 250 мл и вливают 50 мл дистиллированной воды. Колбу Кьельдаля многократно промывают небольшим количеством воды и промывные воды сливают в колбу с раствором. Затем эту колбу закрывают пробкой, имеющей два отверстия— для капельной воронки и для каплеуловителя (см. рис. 45, б). Каплеуловитель соединяют с холодильником, конец которого помещен в коническую колбу (приемник), содержащую 25 мл 1 н. раствора серной кислоты. В капельную воронку наливают 100 мл 40%-ного раствора гидроксида натрия и медленно приливают его в перегонную колбу. Колбу нагревают вначале интенсивно, чтобы быстро довести ее содержимое до кипения, а затем умеренно, до тех пор, пока не будет отогнано около 2/з жидкости, и проверяют красной лакмусовой бумагой отсутствие аммиака в капле дистиллята. По окончании перегонки промывают холодильник дистиллированной водой и избыток кислоты титруют 1 н. раствором гидроксида натрия в присутствии индикатора метилового оранжевого. По количеству выделившегося аммиака вычисляют содержание азота в анализируемом продукте. Массовую доля азота ω (%) рассчитывают по формуле
где K1 — коэффициент поправки для 1 н. раствора серной кислоты; К2— коэффициент поправки для 1 н. раствора гидроксида натрия; V— объем 1 н. раствора гидроксида натрия, израсходованный на титрование избытка кислоты; т — масса навески акрилонитрила; 0,014 — масса азота, соответствующая 1 см3 точно 1 н. раствора серной кислоты, г. На основе полученных данных можно вычислить массовую долю акрилонитрила (%), умножив массовую долю азота (%) на фактор пересчета, равный 3,786. Работа 10. Определение массовой доли акрилонитрилапотенциометрическим методом.Метод основан на взаимодействии акрилонитрила с сульфитом натрия:
Выделяющаяся в результате реакции щелочь потенциометрически оттитровывается соляной кислотой. Молярная масса эквивалента акрилонитрила равна массе его моля. Реактивы и оборудование Диоксан Сульфит натрия, 0,5 н. раствор Соляная кислота, 0,1 и. раствор Бюкс для взятия навески Колба коническая на 250 мл с пришлифованной пробкой Пипетка на 25 мл Установка для потенциометрического титрования
Ход определения. Навеску акрилонитрила ~1 г, взятую с точностью до 0,002 г, вносят в коническую колбу с притерной пробкой емкостью 250 мл , растворяют в 25 мл диоксана и приливают пипеткой 25 мл 0,5 н. раствора сульфита натрия. Содержимое колбы перемешивают и через 30 мин выделившуюся щелочь потенциометрически титруют 0,1 н. раствором соляной кислоты. Перед титрование анализируемой пробы замеряют потенциал стеклянного электрода в контрольном растворе. Для этого в стакан вносят 25 мл 0,5 н. раствора сульфита натрия и 150 мл дистиллированной воды. В раствор опускают стеклянный и каломельные электроды, включают мешалку и замеряют на потенциометре значение потенциала стеклянного электрода (мВ). Затем контрольный раствор выливают и электроды тщательно промывают дистиллированной водой. Анализируемую пробу количественно переносят в чистый стакан, приливают 125 мл дистиллированной воды и погружают электроды. Титрование ведут до значения потенциала стеклянного электрода в контрольном растворе, после чего отмечают объем раствора соляной кислоты, пошедший на титрование анализируемой пробы. Массовую долю акрилонитрила в ω (%) вычисляют по формуле
где V—объем 0,1 н. раствора соляной кислоты, затраченный на титрование анализируемой пробы; К. — коэффициент поправки для 0,1 н. раствора соляной кислоты; 0,0053 — масса акрилонитрила, соответствующая 1 см3 точно 0,1 н. раствора соляной кислоты, г; т — навеска акрилонитрила.
ИНИЦИАТОРЫ
В качестве инициаторов радикальной полимеризации применяют перекисные соединения, такие, как пероксида бензоила и водорода, персульфаты и др. При нагревании пероксиды разлагаются с образованием свободных радикалов, которые становятся источником роста полимерной цепи. Пероксид бензоила(С6Н5СОО)2 (Мr =242,16) Белые кристаллы без запаха. Не растворима в воде, в спирте, растворима во многих органических растворителях, мономерах. По ГОСТ 14888—78 массовая доля пероксида бензоила в сухом продукте не менее 98%. Период полураспада 13 ч при 70°С и 24 мин при 100°С. Сухой пероксид бензоила взрывается при трении и ударе. Бурно разлагается при температуре плавления 101°С. При работе с пероксидом бензоила необходимо строго соблюдать меры предосторожности.
Работа 1. Определение массовой доли пероксида бензоила.Количественное определение основано на способности пероксида бензоила в кислой среде выделять иод из иодида калия:
Выделяющийся в результате реакции иод оттитровывают раствором тиосульфата натрия:
Молярная масса эквивалента пероксида бензоила равна 1/2 массе его моля.
Ход определения. Навеску перекиси бензоила ~0,2 г, взятую с точностью до 0,0002 г, растворяют в конической колбе в 10 мл ацетона, добавляют 3 мл насыщенного раствора иодида калия и подкисляют несколькими каплями уксусной кислоты. Через 20 — 25 мин раствор разбавляют 15 мл воды и оттитровывают выделившийся иод 0,1 н. раствором тиосульфата натрия в присутствии крахмала, добавляемого в конце титрования. Параллельно в тех условиях проводят контрольный опыт. Массовую долю пероксида бепзоила ω (%) вычисляют по формуле
где v1— объем 0,1 н. раствора тиосульфата натрия, израсходованный на титрование пробы; v2— объем 0,1 н. раствора тиосульфата натрия, израсходованный на титрование в контрольном опыте; К. — коэффициент поправки для 0,1 н. раствора тиосульфата натрия; 0,01211 — масса пероксида бензоила, соответствующая 1 см3 точно 0,1 н. раствора тиосульфата натрия, г; т — навеска пероксида бензоила. Пероксид водородаНО— ОН(Мr= 34,02) Бесцветная жидкость, смешивается с водой во всех отношениях. Выпускается в виде 30%-ного (пергидроль) раствора в воде (ГОСТ 177—77).
Персульфат аммония(NН4)2S2О8 (Мr=228,21) Белые кристаллы. Плотность 1,98 г/см3. Температура разложения 120°С. Сухая соль сохраняется без разложения неограниченное время, влажная — постепенно разлагается, выделяя кислород и озон. В водных растворах разлагается уже при комнатной температуре и особенно при нагревании (ГОСТ 20478—75). Определение массовой доли персульфата аммония.Количественное определение основано на способности персульфата аммония окислять железо (II) в железо (III) по уравнению реакции
Избыток сульфата железа (II) оттитровывают раствором перманганата калия:
Молярная масса эквивалента персульфата аммония равна 1/2 массы его моля. Ход определения. Взвешивают ~0,3 г персульфата аммония с точностью до 0,0002 г, помещают его в коническую колбу емкостью 250 мл, растворяют его в 10—15 мл воды и добавляют цилиндром 5 мл 25%-ного раствора серной кислоты и пипеткой 30 мл 0,2 н. раствора соли Мора. Затем приливают 100 мл воды, нагретой до 70—80°С, и избыток соли Мора титруют 0,1 и. раствором перманганата калия. Параллельно проводят контрольный опыт. Массовую долю персульфата аммония ω (%) вычисляют по формуле
где v1— объем 0,1 н. раствора перманганата калия, израсходованный на титрование контрольного опыта; v2— объем 0,1 н. раствора перманганата калия, израсходованный на титрование пробы; К. — коэффициент поправки для 0,1 н. раствора перманганата калия; 0,01141 — масса персульфата аммония, соответствующая 1 см3 точно 0,1 н. раствора перманганата калия, г; т — масса персульфатааммония. Персульфат калияК2S2О8 (Мr= 270,29) Белые кристаллы. Плотность 2,48 г/см3. Температура разложения около 100°С. При нагревании в водных растворах распадается на радикалы, инициирующие полимеризацию. ГОСТ 4146—74.
Работа 4. Определение содержания персульфата калия.Количественное определение основано на способности персульфата калия в кислой среде выделять иод из раствора иодида калия:
Выделившийся в результате реакции иод оттитровывают раствором тиосульфата натрия:
Молярная масса эквивалента персульфата калия равна 1/2 массы его моля.
Реактивы и оборудование
Иодид калия, 10%-иый раствор Колбы конические на 250 мл с при- Серная кислота, 25%-ный раствор шлифованной пробкой Тиосульфат натрия, 0,1 н. раствор Мерные цилиндры на 10 и 50 мл Крахмал, 1%-ный раствор
Ход определения. Около 0,3 г персульфата калия, взвешенного с точностью до 0,0002 г, растворяют в конической колбе емкостью 250 мл в 20 — 30 мл воды, добавляют 10 мл 10%-ного раствора иодида калия и 15 мл 25%-ного раствора серной кислоты. Иодид калия и серную кислоту добавляют цилиндром. Колбу закрывают пробкой и оставляют стоять на 2 ч. Затем выделившийся иод оттитровывают 0,1 н. раствором тиосульфата натрия в присутствии крахмала, который добавляют в конце титрования, когда цвет раствора станет соломенно-желтым. Параллельно проводят контрольный опыт. Массовую долю персульфата калия ω (%) вычисляют по формуле
где v1— объем 0,1 н. раствора тиосульфата натрия, израсходованный на титрование пробы; V2 — объем 0,1 н. раствора тиосульфата натрия, израсходованный на титрование контрольного опыта; K — коэффициент поправочный для 0,1 н. раствора тиосульфата натрия; 0,013516 — масса персульфата калия, соответствующая 1 см3 точно 0,1 н. раствора тиосульфата натрия, г; т — масса навески персульфата калия.
ПЛАСТИФИКАТОРЫ В промышленности пластических масс применяют многие пластификаторы, принадлежащие к различным классам органических соединений. Наиболее распространены пластификаторы эфирного типа: эфиры многоосновных кислот и одноатомных, спиртов, эфиры одноосновных кислот и многоатомных спиртов, эфиры жирных кислот. В качестве пластификаторов часто применяют производные фосфорной кислоты — ее эфиры с низшими спиртами, хлорпроизводпые эфиров, эфиры с фенолами, а также азотсодержащие соединения (производные мочевины), хлорпроизводные (хлорированные дифенилы, хлорированные парафины), камфару, ацетофенон и др. Жидкие пластификаторы (ГОСТ 8728—77) характеризуют по плотности, температуре вспышки, кислотному числу, числу омыления. Твердые пластификаторы характеризуют по плотности, температуре плавления, растворимости. Определение кислотного числа и числа омыления в пластификаторах эфирного типа.Кислотное число показывает массу гидроксида калия, необходимую для нейтрализации свободных кислот, содержащихся в 1 г эфира. Ход определения. Навеску пластификатора ~10 г, взятую с точностью до 0,0002 г, помещают в коническую колбу емкостью 250 мл, растворяют в 50 мл этилового спирта и титруют полученный раствор 0,1 н. раствором гидроксида калия в присутствии фенолфталеина до появления светло-розовой • окраски. Кислотное число вычисляют по формуле
где V— объем 0,1 н. раствора гидроксида калия, израсходованный на титрование пробы; К-—коэффициент поправки для 0,1 н. раствора гидроксида калия; т — масса навески пробы; 5,6—масса гидроксида калия в 1 см3 0,1 н. раствора, мг. Число омыления показывает, какая масса гидроксида калия необходима для нейтрализации свободных и связанных кислот, содержащихся в 1 г эфира. Для определения числа омыления пластификатор обрабатывают избытком спиртового раствора щелочи до полного омыления и титруют избыток щелочи раствором кислоты в присутствии фенолфталеина. Ход определения. Навеску ~ 1 г пластификатора, взятую с точностью до 0,0002 г, помещают в коническую колбу емкостью 250 мл; приливают пипеткой 50 мл 0,5 п. раствора гид-роксида калия в этиловом спирте, присоединяют к колбе обратный холодильник и нагревают на водяной бане в течение 2 ч. Охлаждают и титруют 0,5 и. раствором соляной кислоты в присутствии фенолфталеина до исчезновения розовой окраски. Одновременно в тех же условиях проводят контрольный опыт. Число омыления (ч.о.) вычисляют по формуле
где v1— объем 0,5 п. раствора соляной кислоты, израсходованный на титрование в контрольном опыте; V2— объем 0,5 п. раствора соляной кислоты, израсходованный на титрование пробы; К — коэффициент поправки для 0,5 п. раствора соляной кислоты; т — масса навески пластификатора; 28,05 — масса гид-роксида калия, соответствующая 1 см3 0,5 н. раствора соляной кислоты, мг.
ТЕХНИЧЕСКАЯ ВОДА
Природные воды бывают подземные и поверхностные: речные, озерные, воды морей и океанов, а также дождевая вода. Являясь хорошим растворителем, вода содержит различные примеси, от концентрации и природы которых зависит ее пригодность для промышленных и бытовых целей. Значительное влияние на состав поверхностных вод оказывают сточные воды населенных пунктов и отходы промышленных производств. В зависимости от использования воды к ней предъявляют требования,которые устанавливаются государственными стандартами (ГОСТ). Если вода, поступающая из водоисточника, не удовлетворяет требованиям соответствующего ГОСТа, проводят водоподготовку. Питьевая вода должна отвечать нормам ГОСТ 2874—82, где предусмотрены прежде всего санитарные требования. Воду широко применяют во всех отраслях промышленности для самых различных целей: как теплоноситель — для подачи тепла и отвода его; в качестве рабочего тела для передачи работы в гидропрессах; для транспортировки полупродуктов химических производств; как технологический компонент в различных производственных процессах. Требования к качеству воды для разных отраслей промышленности сводятся в основном к условию: примеси не должны вредить ее производственному использованию. Вода не должна вызывать коррозию котлов, аппаратуры, труб; она не должна содержать избытка взвешенных веществ, забивающих трубки охлаждающей системы, засорять и истирать детали прессов. Соли, вызывающие жесткость воды, и другие примеси образуют на рабочих поверхностях теплосиловых установок низкотеплопроводпые отложения (накипи). При этом ухудшается теплообмен в аппаратах, происходит перерасход топлива, повреждается поверхность металла. Вода, поступающая из водоисточников, подвергается анализу до и после водоподготовки. При полном анализе определяют содержание взвешенных веществ, сухой остаток, жесткость, окисляемость, щелочность, кислотность, содержание различных ионов (Са2+, Мg2+, Fе2+, Fе3+, НСО3- и др.). Отбор пробы при исследовании любых компонентов воды является одним из важнейших моментов. Прежде всего необходимо обеспечить полнейшую чистоту склянок, применяемых для отбора пробы воды. Рекомендуется пользоваться склянками из бесцветного стекла с притертыми пробками, которые предварительно многократно ополаскивают исследуемой водой. При взятии пробы водопроводной воды ее предварительно спускают, чтобы в склянку не попала застоявшаяся в трубах вода. Методы отбора проб воды указаны в ГОСТ 24481—-80 и ГОСТ 18963—73. Для проведения полного анализа необходимо иметь около пяти литров воды.
Определение сухого остатка воды (ГОСТ 18164—72).Твердые вещества, остающиеся после упаривания воды, называют сухим остатком. Следует обратить внимание, что цвет сухого остатка должен быть чисто-белым. Воды, содержащие органические загрязнения, дают темный сухой остаток. Если сухой остаток обугливается, вода сильно загрязнена. Обычно в воде общее количество растворенных веществ не превышает 500 мг/л. Содержание сухого остатка в исследуемой воде выражают в миллиграммах на литр воды.
Оборудование Чашка фарфоровая на 100 мл Эксикатор Колба мерная на 200 мл
Ход определения. Для определения сухого остатка берут в мерную колбу 200 мл воды. Упаривают воду па водяной бане в предварительно прокаленной и взвешенной фарфоровой чаише, постепенно доливая до тех пор, пока вся рода не будет переведена в выпарную чашку. Затем мерную колбу ополаскивают дистиллированной водой и эту воду также упаривают. После этого чашку переносят в сушильный шкаф, сушат в течение трех часов при 110°С, охлаждают в эксикаторе и взвешивают. Взвешивание должно проводиться тотчас после того, как чашка вынута из эксикатора, так как сухой остаток иногда притягивает воду. Массовую концентрацию сухого остатка с (мг/л) определяют по формуле
где т — масса чашки с сухим остатком; т1— первоначальная масса чашки; V—объем анализируемой воды. Жесткость воды определяется содержанием в пей солей (обычно кальция и магния). Различают карбонатную, некарбонатную и общую жесткость. Карбонатная жесткость обусловливается содержанием карбонатов кальция и магния; некарбонатная жесткость — содержанием других кроме гидрокарбонатов солей кальция и магния (хлоридов, сульфатов); общая жесткость — общим содержанием солей. При длительном кипячении воды гидрокарбонаты выделяют двуоксид углерода и переходят в карбонаты, выпадающие в осадок, вследствие чего жесткость уменьшается:
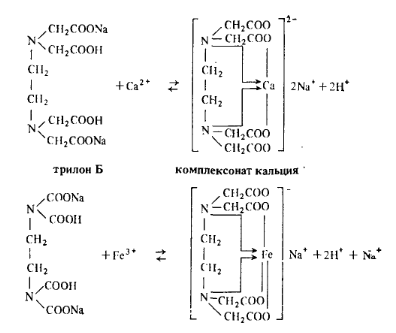
Кипячением карбонатная жесткость полностью не устраняется, поэтому иногда применяют термин, устранимая жесткость, которым определяют концентрацию бикарбонатов, удаляемых из воды при кипячении ее в течение 1 ч. Оставшуюся после кипячения воды жесткость называют постоянной. В СССР жесткость воды выражают в миллимолях соли на 1 л воды. 1 млмоль Са2+ = 20,04 мг, 1 млмоль Мg2+= 12,16 мг. Жесткость природной воды колеблется в широких пределах. Вода с жесткостью менее 4 млмоль/л характеризуется как мягкая, от 4 до 8 млмоль/л — средняя жесткость, от 8 до 12 млмоль/ л — жесткая и выше 12 млмоль/л — очень жесткая. Определение общей жесткости воды комплексо-метрическим методом (ГОСТ 4151—72).Комплексометрический метод основан па способности ряда катионов образовывать достаточно прочные растворимые внутрикомплексные соединения с комплексонами —органическими веществами, являющимися производными иминодиуксусной кислоты NН(СН2СООН)2. Наибольшее распространение в комплексометрии получил трилон Б (титрованный раствор двузамещенной натриевой соли этилепдиаминтетрауксусной кислоты), дающей комплексные соединения с катионами двух- и трехвалентных металлов: Формулу трилона Б можно изобразить Nа2Н2R, где Н2R2 — анион комплексона.
Из приведенных схем реакций видно, что 1 моль трилона Б всегда связывает 1 моль металла независимо от его валентности. При этом освобождается 2 моль водорода, которые могут быть оттитрованы в присутствии кислотно-основного индикатора. Комплексные соединения, образуемые трилоном Б с различными ионами, называют комплексонатами. Комплексонаты обладают различной прочностью, которую принято выражать количественно значением константы нестойкостиКн, или константой диссоциации комплексного иона. Чем меньше ее значение, тем устойчивее комплекс. Удобнее прочность комплексного соединения выражать величиной, равной отрицательному логарифму константы нестойкости рК.. При этом чем больше значение р/(, тем выше прочность соответствующего комплекса. Значения рК. зависят от рН раствора и температуры. Сопутствующие ионы, имеющие большее значение рК, чем определяемый ион, а следовательно, дающие более прочные комплексы, будут мешать при титровании анализируемого раствора трилоном Б. Ионы, мешающие определению анализируемого катиона, должны быть удалены из сферы реакции осаждением или маскированы переводом в бесцветные комплексы, более устойчивые, чем комплексонаты. Для маскировки катионов применяют гидроксиламин (Мn2+), триэтаноламин (Fе2+, А13+, Мn2+) и др. В качестве индикаторов при комплексометрическом титровании применяют различные органические вещества, в основном красители, Образующие комплексы с ионами определяемых металлов (металлинипдикаторы). Наибольшее распространение получили индикаторы: кислотный хром темно-синий, хромоген черный ЕТ-00, мурексид. Хромоген черный ЕТ-00 реагирует с катионами многих металлов. Сокращенная формула этого индикатора NаН2Ind, где Ind — анион индикатора. При рН<6 он имеет виппо-красный цвет, при рН 7÷11 — синий, при рН>>12— желто-оранжевый. С кальцием, магнием, цинком, кадмием и свинцом этот индикатор образует окрашенные в винно-красный цвет устойчивые комплексы. Прочность металлинипдикатора меньше прочности соответствующего комплексопата. Окраска металлининдикатора отличается от окраски самого индикатора. При добавлении к анализируемому раствору титрованного раствора трилона Б в эквивалентной точке, когда полностью разрушается за счет образования комплексоната металлининдикатор, появляется окраска самого индикатора. Например, при определении кальция протекает следующая реакция:
Трилонометрическое определение каждого иона производят при том значении рН, при котором этот ион образует с трилоном Б соединение более прочное, чем с индикатором. Для поддержания заданного рП при определении того или иного катиона к титруемому раствору добавляются буферные растворы. Кроме того, поддержание заданной величины рН обеспечивает определенную окраску индикатора. Жесткость воды определяют при рН>9. Приготовление реактивов. 1. Титрованный раствор трилона Б. При жесткости воды выше 20 млмоль/л титрование пробы производят 0,1 н. раствором трилона Б, при жесткости 0,5—20 млмоль/л — 0,05 н. раствором и при жесткости ниже 0,5 млмоль/л—0,01 н. раствором. Для приготовления растворов трилона Б берут следующие его количества: для 0,1 н. раствора—18,6 г трилона Б, для 0,05 н.—9,3 г и для 0,01 к.— 1,86 г. Навеску трилона Б растворяют в дистиллированной воде и фильтруют, если раствор получится мутным. Объем раствора доводят до 1 л. Для приготовления рабочих растворов и разбавления проб необходима высокочистая дистиллированная вода. Вода считается пригодной, если после прибавления к 100 мл дистиллята 1 мл аммиачного буферного раствора и 5—7 капель индикатора кислотного хрома темно-синего окраска воды будет голубая с сиреневым оттенком. Для установки титров растворов трилона Б разной концентрации берут различные объемы 0,01 н. раствора сульфата магния, приготовленного из диксанала. Для 0,1 н. раствора трилона Б нужно 100 мл раствора MgSO4, для 0,05 н. раствора — 50 мл и для 0,01 н.— 10 мл. Объем взятого раствора соли магния доводят дистиллированной водой до 100 мл, добавляют 5 мл аммиачного буферного раствора, 5—7 капель индикатора кислотного хрома темно-синего и медленно титруют раствором трилоиа Б при перемешивании до изменения окраски раствора. Коэффициент поправки для приведения раствора трилона Б к данной молярной концентрации эквивалента (нормальность) вычисляют но формуле где V—расход трилона Б; KМg — поправочный коэффициент соли магния (при точно 0,01 н. растворе KМg=1). 2. Приготовление буферного раствора: 20 г хлорида аммония растворяют в дистиллированной воде, добавляют 100 мл 25%-ного раствора аммиака и доводят до 1 л дистиллированной водой. 3. Приготовление растворов индикаторов: хромоген черный ЕТ-00 и кислотный хром темно-синий готовят растворением 0,5 г индикатора в 20 мл аммиачного буферного раствора и доводят до 100 мл этиловым спиртом. Раствор хромогена черного ЕТ-00 можно хранить не более 10 сут. Мурексид готовят растворением 0,03 г его в 10 мл дистиллированной воды. Раствор хранят в темном месте, срок годности 4 сут. Реактивы и оборудование
Сульфид натрия, 5%-ный раствор Гидроксиламин хлорид, 1%-ный раствор Трилон Б Буферный раствор Индикаторы Пипетка на 100 мл Колба коническая на 250 мл Микробюретка
Ход определения. 1. При отсутствии ионов меди, цинка, марганца. Для определения общей жесткости воды отмеривают пипеткой 100 мл анализируемой прозрачной пробы воды, переносят ее в коническую колбу емкостью 250 мл, добавляют 5 мл аммиачного буферного раствора и 5— 7 капель индикатора хромогена черного ЕТ-00. После тщательного перемешивания смесь окрашивается в винно-красный цвет. Пробу титруют раствором трилона Б из микробюретки до изменения окраски. Титрование проводят медленно, непрерывно перемешивая анализируемую пробу воды. Жесткость воды Жобщ(млмоль/л) рассчитывают по формуле где v1— объем пробы воды; с — молярная концентрация эквивалента раствора трилона Б; V2—-объем раствора трилона Б, израсходованный на титрование; К — коэффициент поправки для приведения раствора трилона Б к данной концентрации. 2. В присутствии ионов меди и цинка. Отмеривают пипеткой 100 мл пробы воды и помещают ее в коническую колбу емкостью 250 мл. Затем прибавляют 1 мл свежеприготовленного 5%-ного раствора сульфида натрия. При этом выпадают в осадок СиЗ и 2п5. Добавив 5 мл аммиачного буферного раствора, 5—7 капель индикатора хромогена черного ЕТ-00, титруют пробу воды раствором трилона Б. Расчет выполняют как в предыдущем анализе. 3. В присутствии ионов марганца. К 100 мл воды, взятой для определения жесткости, добавляют 3 капли раствора гидроксиламина хлорида (1 г МН4ОН-НС1 растворяют в дистиллированной воде и доводят до объема 100 мл). При этом происходит маскировка катиона марганца. Затем прибавляют буферный раствор, индикатор и титруют раствором трилона Б соответствующей концентрации. Определение карбонатной, устранимой и некарбонатной жесткости.Карбонатную жесткость Жк, обусловленную присутствием в воде растворимых бикарбонатов кальция и магния, определяют титрованием пробы воды соляной кислотой в присутствии метилового оранжевого:
Устранимую жесткость определяют по разности содержания ионов HСО3до и после кипячения пробы воды, некарбонатную — по разности между общей жесткостью и карбонатной.
Реактивы и оборудование
Соляная кислота, 0,1 и 0,01 н. раст- Пипетка на 100 мл вор Колба коническая на 250 мл Метиловый оранжевый, 0, 1%-ный Колба мерная па 200 мл Раствор
Ход определения. Отмеряют пипеткой пробу воды 100 мл и переносят в коническую колбу емкостью 250 мл, добавляют 2—3 капли метилового оранжевого и титруют 0,1 н. раствором соляной кислоты до перехода желтой окраски в устойчивую оранжевую. При этом определяют общую карбонатную жесткость. Расчет жесткости (млмоль/л) проводят по формуле
где V— расход 0,1 н. соляной кислоты; K — коэффициент поправки для 0,1 н. раствора соляной кислоты. Затем определяют остаточную карбонатную жесткость. Пробу воды 200 мл кипятят в конической колбе в течение 1 ч. После охлаждения воду фильтруют, отбирают 100 мл фильтрата в коническую колбу и титруют 0,01 н. раствором соляной кислоты в присутствии метилового оранжевого. Остаточную жесткость Жост (млмоль/л) рассчитывают по формуле
где V— расход 0,01 н. раствора соляной кислоты; К — коэффициент поправки для 0,01 н. раствора соляной кислоты. По разности между всей карбонатной и остаточной жесткостью находят устранимую жесткость: жу = жк — жост Методом комплексометрии определяют общую жесткость и по разности между общей и карбонатной жесткостью вычисляют некарбонатную жесткость. Определение окисляемости воды. Косвенным показателем содержания в сточной воде органических веществ является ее окисляемость, показывающая расход кислорода (или перманганата калия) на окисление органических веществ в воде. Принцип метода основан па том, что кислый или щелочной раствор перманганата способен отдавать кислород в присутствии окисляющихся веществ. С целью учета вошедшего в реакцию кислорода избыток перманганата восстанавливается щавелевой кислотой:
Окисляемость выражают в миллиграммах кислорода или перманганата калия на 1 л воды.
Реактивы и оборудование
Серная кислота, 25%-ный раствор Колба коническая на 250 мл Перманганат калия, 0,01 н. раствор Пипетка градуированная на 10 мл Щавелевая кислота, 0,01 н. раствор Цилиндр мерный на 10 мл
Ход определения. В коническую колбу емкостью250мл отмеривают пипеткой от 1 до 20 мл исследуемой воды, доливают дистиллированной водой до 100 мл и прибавляют 5 мл 25%-ного раствора серной кислоты. Раствор доводят до кипения. Затем прибавляют из бюретки 10 мл 0,01 н. раствора перманганата калия и капитят ровно 10 мин с момента появления первого пузырька воздуха. После кипячения жидкость должна иметь красную окраску, что указывает на избыток окислителя. В случае исчезновения окраски следует повторить определение, взяв для анализа меньшее количество воды. К горячей жидкости приливают из бюретки 10 мл 0,01 н. раствора щавелевой кислоты, при этом раствор обесцвечивается. Избыток щавелевой кислоты оттитровывают 0,01 н. раствором перманганата калия до появления розовой окраски. Одновременно проводят контрольный опыт, для чего в колбу наливают 5 мл 25%-ного раствора серной кислоты, 100 мл дистиллированной воды и далее определение проводят так же, как и для исследуемой воды. Расчет окисляемости ведут по формуле где V3— объем 0,01 п. раствора перманганата калия, принятый для определения в основном и контрольном опытах; v1— объем 0,01 н. раствора пермангапата калия, израсходованный на титрование избыточного раствора щавелевой кислоты в основном опыте; v2— объем 0,01 п. раствора перманганата калия, израсходованный на титрование в контрольном опыте; К — коэффициент поправки для 0,01 п. раствора пермангапата калия; 0,08 — масса кислорода, соответствующая 1 мл точно 0,01 н. раствора пермапгаиата калия, г; V— объем исследуемой воды. Следует, однако, иметь в виду, что потребление перманганата калия водой не является абсолютной мерой содержания в ней органических веществ, так как различные органические вещества потребляют различное количество перманганата калия, а кроме того, почти никогда не наблюдается полного их окисления. Работа 5. Определение содержания железа. В природных водах железо содержится в небольших количествах, в основном в виде гидрокарбопата Fе(НСО3)2. В результате действия кислорода частьFе2+ переходит в Fе3+. Железо определяют колориметрическими методами, например роданидным методом. В основе роданидного метода лежит свойство иона железа Fе3+ образовать с ионами роданида в зависимости от концентрации последних ряд комплексных ионов кроваво-красного цвета, обусловливающих различную интенсивность окраски раствора:
где п— число ионов роданида, связанных в тиоционатный комплекс (п может меняться от 1 до 6). Определению железа в виде роданида мешают ионы, образующие комплексные соединения с ионами Ре3+; восстановители, восстанавливающие Ре3+ до Fе2+; окислители, разрушающие роданид-ион. Данным методом можно определить содержание только Fе3+, так как Fе2+ с роданидом не дает окрашенных соединений. Для определения общего содержания железа ионы Fе2+предварительно окисляют в ионы Fе3+. Чувствительность метода равна 2,5 мкг железа в 50 мл раствора при толщине слоя окрашенного раствора 5 см.
Реактивы и оборудование Эталонный раствор соли железа Фотоэлектроколориметр ФЭК-М Соляная кислота, р=1,10 Колбы мерные на 50 мл — 6 шт. Роданид аммония, 10%-ный раствор Пипетка градуированная на 5 мл Персульфат аммония Пипетка на 25 мл Приготовление эталонного раствора соли железа. Навеску 0,864 г железоаммонийных квасцов NН4Fе(SО4)2· 12Н2О (х.ч.) растворяют в дистиллированной воде, подкисляют 5 мл серной кислоты (р=1,84) и доводят объем раствора водой до 1 л. Раствор содержит 0,1 мг железа в 1 мл. Ход определения. Перед определением предварительно строят градуировочную кривую D=f(с) по эталонному раствору железа, выражающую зависимость оптической плотности окрашенного раствора от его концентрации. Для построения графика готовят в шести мерных колбах емкостью по 50 мл растворы соли железа разной концентрации. Наливают последовательно 0,5; 1,0; 2,0; 3,0; 4,0; 5,0 мл эталонного раствора соли железа, добавляют в каждую колбу по 1 мл разбавленной (1:1) соляной кислоты и по 5 мл 10%-ного раствора роданида аммония (или калия). Во всех колбах объем растворов доводят до метки и измеряют оптические плотности растворов с синим светофильтром в кюветах с толщиной слоя 1 см на фотоэлектроколориметре ФЭК-М. Оптическую плотность каждого раствора измеряют 4—5 раз и вычисляют средние арифметические значения, которые откладывают по оси ординат при построении градуировочной кривой; по оси абсцисс откладывают концентрацию. Вследствие сравнительно быстрого разрушения окраски колориметрического раствора роданид аммония следует приливать в колбу непосредственно перед измерением. После этого колориметрируют пробу воды. Для этого берут 25 мл пробы и переносят в колбу емкостью 50 мл. Приливают 1 мл разбавленной (1:1) соляной кислоты и вносят 2—3 кристалла персульфата аммониня для окисления Fе2+ в Fе3+. Раствор нагревают до кипения на водяной бане, выдерживают 10 мин и охлаждают. Затем в колбу добавляют 5 мл 10%-ного раствора роданида аммония. При этом исследуемая проба воды окрашивается в красный цвет. Получив среднеарифметическое значение оптической плотности из 4—5 измерений, находят это значение па ординате графика, проводят из этой точки горизонтальную линию до пересечения с градуировочной кривой и опускают перпендикуляр на ось абсцисс. Точка пересечения с абсциссой дает значение искомой концентрации железа в пробе воды.
ГЛАВА 8
|
||
|
|
Последнее изменение этой страницы: 2018-05-10; просмотров: 468. stydopedya.ru не претендует на авторское право материалов, которые вылажены, но предоставляет бесплатный доступ к ним. В случае нарушения авторского права или персональных данных напишите сюда... |