|
Студопедия КАТЕГОРИИ: АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция |
Доминантные и рецессивные признаки у человека 1 страница(для некоторых признаков указаны контролирующие их гены)
Неполное доминирование(указаны гены, контролирующие признак)
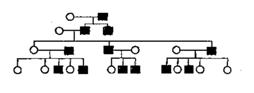
58 Методы изучения наследственности человека. Их достоинства и возможности. Клинико-генеалогический, близнецовый, популяционно-статистический, цитогенетический, биохимический, иммунологический методы. Методы рекомбинантной ДНК и гибридизации соматических клеток. Биологическое и математическое моделирование. Пренатальная (дородовая) диагностика. Дерматоглифика. 1.Генеалогический метод.  -С помощью генеалогического метода можно выяснить характер наследования : доминантный или рецессивный, аутосомный или сцепленный с полом. Если признак является доминантным аутосомным, то в родословной как правило, такой признак проявляется в каждом поколении, его обладателями в равной степени являются мужчины и женщины (рис.2) Рис 2. Родословная с аутосомно-доминантным наследованием
Рецессивный признак проявляется у гетерозиготных обладателей рецессивных генов, поэтому такие признаки проявляются не в каждом поколении и вероятность их выщепления значительно возрастает в родственных браках, что является научным основанием для их запрета. Среди мужчин и женщин рецессивные аутосомные признаки встречаются с равной частотой (рис.3).
Сцепленные с Х-хромосомой рецессивные признаки проявляются, как правило, чаще у мужчин, так как для этого достаточно одной дозы гена в Х- хромосоме. Особь, у которой рецессивный ген проявляется в одной дозе, называется гемизиготой. У женщин такие признаки проявляются значительно реже, так как для этого нужно, чтобы обе Х-хромосомы несли рецессивные
Признаки, которые передаются через У-хромосому, называются голандрическими. Они передаются и проявляются только у мужчин (рис.5)
Близнецовый метод. В настоящее время известно, что способность к рождению ОБ имеет генетическую обусловленность и передается по наследству. Такие близнецы представляют большой интерес для изучения роли наследственности и среды в формировании признаков человека, в том числе и таких сложных, как психические. Было сделано интересное наблюдение : у разлученных ОБ, очень часто наблюдается удивительное сходство не только морфологических, но и психологических особенностей: сходные пристрастия, приоритеты, профессии, семейный уклад . Был сделан вывод о том , что «сходные генотипы выбирают сходную среду»
Если близнецы появляются из разных, но одновременно овулирующих и оплодотворенных яйцеклеток, то их называют разнояйцевыми (РБ). Разнояйцевые близнецы похожи не более, чем дети из одной семьи. Они могут быть одного или разного пола. Рождение РБ связано с нарушением образования женских половых клеток – овогенеза, и часто это происходит у женщин более зрелого возраста. РБ также представляют определенный интерес для исследователя-генетика. В этом случае исследователь исходит из посыла, что близнецы имеют разный генотип, но средовые факторы на них действуют одинаковые (общая внутриутробная среда до рождения, одинаковые семейные условия, одинаковая, как правило, школьная среда и т.п.). В этом случае, все фенотипические различия между РБ предписываются генетическим факторам.
Цитогенетический метод Этот метод стали применять с 60-ых годов ХХ века, после установления в 1956 году Дж. Тийо и А.Леваном точного числа хромосом человека. На Международных совещаниях в Денвере (1960), Лондоне (1963), Чикаго (1966) была разработана классификация и номенклатура хромосом человека . В соответствии с рекомендациями этих конференций хромосомы располагаются в порядке уменьшения их длины. Все пары аутосом пронумерованы арабскими цифрами с 1(самая крупная) до 22( самая мелкая) и 23 пара – половые хромосомы : ХХ или ХУ По положению центромеры различают: равноплечие хромосомы (метацентрические), слабонеравноплечие (субметацентрические), сильнонеравноплечие (акроцентрические) В соответствии с морфологическими особенностями и размером хромосомы человека разделены на семь групп, которые обозначены буквами английского алфавита от А до G. Для дифференцировки хромосом со сходной морфологией и размерами используют специальную их обработку – дифференциальное окрашивание, в результате которого появляется специфический линейный рисунок по длине хромосомы, связанный с наличием по-разному окрашенных сегментов, уникальных для каждой хромосомы Препараты для анализа кариотипа готовят из делящихся клеток лейкоцитов периферической крови. Разработка методики приготовления и анализа хромосом ознаменовала возникновение новой отрасли исследований – клинической цитогенетики (более подробно о хромосомах см. гл. «Наследственный аппарат соматических и генеративных клеток человека»)
Биохимический метод Механизм развития многих генных заболеваний человека связан с нарушением тем или иным обменом веществ, сопровождающихся появлением в организме повышенных концентраций определенных метаболитов. Молекулярно-генетические методы. Они связаны с выделением молекул ДНК из отдельных хромосом, либо митохондрий, с последующим изучением структуры этих молекул, выявлении изменений в определенных участках гена. Это позволяет проводить молекулярную диагностику наследственной патологии. Полученные этими методами данные позволяют получить более полные представления о геноме человека. Популяционно- статистический метод и генетика популяций Смысл этого закона заключается в том, что если на панмиктическую популяцию не действуют факторы, изменяющие частоты генов, то ее структура остается неизменной в ряду поколений. Установленная закономерность справедлива для идеальной популяции, которая отвечает таким требованиям, как неограниченно большое число особей, что обеспечивает возможность случайного скрещивания; отсутствие мутационного процесса, изменяющего частоты генов; отсутствие оттока генов за счет естественного отбора; отсутствие миграций. Идеальных популяций в природе не существует. В ходе эволюции происходит непрерывная замена одних генотипов другими путем изменения в популяции численного соотношения качественно различающихся генотипов. Равновесие генотипов в панмиктической популяции, изменяется под действием ряда постоянно действующих факторов. К ним относятся мутационный процесс, отбор, изменение численности, изоляция и ряд других. Под действием эндогенных (внутренних) и экзогенных (внешних) факторов происходит непрерывный мутационный процесс. Несмотря на то , что вероятность нарушений ДНК невелика и составляет 1 случай на 100000 – 10000000 нуклеотидов , у человека на одно поколение приходится несколько мутаций структурных генов, и в каждом поколении генофонд популяции пополняется значительным числом новых мутаций. Известно, что у 5% новорожденных регистрируются наследственные заболевания, причем 40% наследственных заболеваний обязаны своим появлением вновь возникшим мутациям. Это объясняется большим число нуклеотидов в геноме человека - 3,2 млрд п.н. Частота аллелей разных генов будет изменяться в зависимости от мутационного давления, т.е от соотношения прямых и обратных мутаций. Мутации составляют первичный источник наследственных изменений для отбора. Отбором - называют процесс переживания организмов, генотипы которых обеспечивают их наибольшую приспособленность к условиям среды. Наиболее эффективным является отбор, направленный против доминантных вредных мутаций. Скорость устранения их значительно выше, чем для рецессивных мутаций, т.к. как правило, они сразу проявляются в фенотипе. Но правила имеют исключения. Так, мутация гена 4 хромосомы приводит к заболеванию хореей Гентингтона, которое характеризуется дегенеративным изменением нервной системы, приводящим к непроизвольным движениям лица и конечностей, затрудненной речи и прогрессирующему слабоумию. Проявляется эта наследственная болезнь в возрасте 35-40 лет, когда больные уже оставили потомков, поэтому такие доминантные гены элиминируются из популяции медленно. Затруднена и элиминация доминантных генов , которые имеют не 100% пенетрантность. (Пенетрантность – способность гена реализоваться в признак) . Так, у человека злокачественная парная опухоль глаз у детей – ретинобластома, обусловлена экспрессией мутантного доминантного гена 13 хромосомы, который имеет пенетрантность 80%. Следовательно 20% генов могут не проявиться и перейти в следующее поколение. Концентрация генов зависит от численности популяций. Рецессивные гены будут устраняться медленнее, поскольку они не проявляются в гетерозиготном состоянии. Чем больше численность популяций, тем меньше вероятность встречи однотипных гетерозиготных генотипов, а значит, меньше вероятность выщепления рецессивных гомозигот. Они начнут выщепляться в гомозиготе лишь тогда, когда концентрация таких генов возрастет. Чем популяция меньше, тем рецессивные гены распространятся быстрее. Именно поэтому в небольших изолированных популяциях (изолятах) чаще происходит выщепление рецессивных генов, которое являются следствием инбридинга – близкородственного скрещивания. Разные изоляты несут различные концентрации сходных генов. Так на Марианских островах смертность среди местного населения от бокового амиотрофического склероза в 100 раз превышает смертность от этой болезни в других странах. В Южной Панаме большую часть племени кариба куна в одной из провинций составляют альбиносы, которые появляются в каждом поколении. В Швейцарии в одном из селений на реке Роне среди 2200 жителей обнаружено 50 глухонемых и 200 человек с дефектами слуха. Дрейф генов- процессы, изменение частоты генов в популяции в ряду поколений под действием случайных (стохастических) факторов, приводящее, как правило, к снижению наследств, изменчивости популяций. Наиб, отчётливо проявляется при резком сокращении численности популяции в результате стихийных бедствий (лесной пожар, наводнение и др.) Характерная особенность динамики генотипич. структуры популяций под действием Д. г. состоит в усилении процесса гомозиготизации, к-рая нарастает с уменьшением численности популяции. Это нарастание обусловлено тем, что в популяциях ограниченного размера увеличивается частота близкородств. скрещиваний, и в результате заметных случайных колебаний частот отдельных генов происходит закрепление одних аллелей при одновременной утрате других. Однако при некоторых условиях (высокая численность популяций; миграционные процессы, при которых приток и отток генов взаимно уравновешиваются; мутационные процессы, уравновешиваемые отбором и т.п.) природные популяции ведут себя как идеальные. Кроме этого в природных популяциях происходят изменения частот аллелей под действием эволюционных факторов очень медленно, поэтому формула Харди-Вайнберга используется в практической деятельности. Ее применяют для изучения генетической структуры природных популяций, вычисления частот встречаемости интересующих генов. Такая информация представляет определенный и теоретический и практический интерес. В медицинской генетике популяционно-статистический метод используется для изучения частоты нормальных и патологических генов, генотипов и фенотипов в популяциях различных стран, городов, местностей, что позволяет составлять прогноз для следующих поколений, в том числе по наследственным патологиям. На основе данных, полученных популяционно-статистическим методом, возможен ретроспективный анализ эволюции генов и их распространения. Так с помощью популяционного метода удалось выявить геногеографию групп крови по системе Ладштейнера (1-4 группы крови) и объяснить ее происхождение.
Дерматоглифика- наука, которая занимается изучением признаков узоров на коже ладонной стороны кистей и стоп человека. Кожа ладонной стороны кистей имеет сложный рельеф — его образуют гребешки, и потому эту кожу называют «гребневой». Гребешки составляют характерные узоры, уникальные для каждого человека и неизменные в течение всей его жизни. Пренатальная диагностика — комплексная дородовая диагностика с целью обнаружения патологии на стадии внутриутробного развития. Позволяет обнаружить более 98 % плодов ссиндромом Дауна (трисомия 21); трисомии 18 (известной как синдром Эдвардса) около 99,9, %, более 40 % нарушений развития сердца и др. В случае наличия у плода болезни родители при помощи врача-консультанта тщательно взвешивают возможности современной медицины и свои собственные в плане реабилитации ребёнка. В результате семьяпринимает решение о судьбе данного ребёнка и решает вопрос о продолжении вынашивания или о прерывании беременности. К пренатальной диагностике относится и определение отцовства на ранних сроках беременности, а также определение пола плода. 59 Классификация наследственных болезней человека. Генные болезни. Фенотипическое проявление генных мутаций — ферментопатии.
Наследственные заболевания — болезни, унаследовавшие потомок от предка, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации Этиологией наследственных болезней являются мутации. Мутации бывают трех видов:
Геномные мутации Причиной геномных мутаций является изменение числа хромосом в клетке. Они вызывают очень сильные изменения в фенотипе, всегда проявляются в первом поколении. Различают три вида геномных мутаций: 1)Полиплоидия 2)Гетероплоидия 3)Гаплоидия -Полиплоидия – это увеличение числа хромосом в геноме клетки, кратное гаплоидному набору хромосом, например, 3n, 4n, 5n,…,120n. Причиной таких мутаций является разрушение веретена-деления в мейозе гаметогенеза, приводящая к образованию полиплоидных гамет и слиянию их в разных сочетаниях. Есть два вида полиплоидии: 1)четная (4n, 6n, 8n…) 2)нечетная (3n, 7n, 9n…) – не образуют гамет, не размножаются, нет в природе. Полисомия по половым хромосомам Трисомия – Х (синдром Трепло Х) кариотип(47, ХХХ)- известны только у женщин, частота синдрома 1: 700 (0,1%).Нерезкие отклонения в физическом развитии, нарушение функций яичников, преждевременный климакс, снижение интеллекта (у части больных признаки могут не проявляться) Тетрасомия (48, ХХХХ) – приводит к умственной недостаточности в разной степени Пентасомия (49, ХХХХХ) – всегда сопровождается тяжелыми поражениями организма и сознания -Гетероплоидия – это изменение числа отдельных хромосом в геноме клетки, не кратное гаплоидному набору хромосом. Причина – разрушение отдельных нитей веретена-деления, образование гетероплоидных гамет и слияния их в разных сочетаниях. Трисомия-21 (болезнь Дауна) - причина патологии-трисомия по 21 хромосоме. Это самая распространенная из всех аномалий, частота рождения составляет 1:500 (до 40% детей с этой болезнью рождают матери старше 40 лет) – монголоидность, укороченные конечности, микроцефалия, аномалии лица, психическая отсталость, снижение иммунитета, 17% больных умирают в первый год жизни. -Гаплоидия – это уменьшение числа хромосом в геноме клетки в 2 раза. Осуществляется при партеногенезе (образование организма из яйцеклетки без оплодотворения ее сперматозоидом). Люди с такой мутацией бесплодны.
Самые частые мутации – это генные. Один ген мутирует раз в 40 тысяч лет, но генов миллионы, поэтому 5-10% генов - мутантны. Генные болезни — это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Причины генных заболеваний: Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов — белков. Любая мутация гена ведет к изменению структуры или количества белка. Начало любой генной болезни связано с первичным эффектом мутантного аллеля. Основная схема генных болезней включает ряд звеньев: мутантный аллель → измененный первичный продукт → цепь биохимических процессов в клетке → органы → организм В результате мутации гена на молекулярном уровне возможны следующие варианты: · синтез аномального белка; · выработка избыточного количества генного продукта; · отсутствие выработки первичного продукта; · выработка уменьшенного количества нормального первичного продукта. Причиной генных мутаций является изменение последовательности нуклеотидов в ДНК, например, добавки, нехватки или перестановки нуклеотидов. Чаще мутирует рецессивный ген, т.к.он неустойчив к неблагоприятным условиям. Такие мутации не проявляются в первом поколении, а накапливаются в генофонде, образуя резерв наследственной изменчивости. Генные мутации подвергаются репарации, т.е. удалению мутации гена и восстановлению поврежденной ДНК. Такие мутации самые частые и изменяют фенотип незначительно. Не заканчиваясь на молекулярном уровне в первичных звеньях, патогенез генных болезней продолжается на клеточном уровне. При различных болезнях точкой приложения действия мутантного гена могут быть как отдельные структуры клетки — лизосомы, мембраны, митохондрии, пероксисомы, так и органы человека. Клинические проявления генных болезней, тяжесть и скорость их развития зависят от особенностей генотипа организма, возраста больного, условий внешней среды (питание, охлаждение, стрессы, переутомление) и других факторов. Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность. Это означает, что одно и то же фенотипическое проявление болезни может быть обусловлено мутациями в разных генах или разными мутациями внутри одного гена. Впервые гетерогенность наследственных болезней была выявлена С. Н. Давиденковым в 1934 г. Общая частота генных болезней в популяции составляет 1-2 %. Условно частоту генных болезней считают высокой, если она встречается с частотой 1 случай на 10000 новорожденных, средней — 1 на 10000 — 40000 и далее — низкой. Моногенные формы генных заболеваний наследуются в соответствии с законами Г. Менделя. По типу наследования они делятся на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х- или Y-хромосомами Ферментопатии — общее название болезней или патологических состояний, развивающихся вследствие отсутствия или нарушения активности каких-либо ферментов или обусловленные полным отсутствием синтеза ферментов или стойкой функциональной недостаточностью ферментных систем органов и тканей. Наследственные ферментопатии. Генетически детерминированные нарушения обмена веществ вследствие Ф. лежат в основе многих наследственных болезней. При этом может полностью отсутствовать ген, контролирующий синтез белковой молекулы фермента (апофермента), либо апофермент синтезируется, но активность фермента отсутствует или резко снижена. В результате генных мутаций может изменяться последовательность аминокислот в структуре активного центра фермента или в регионе связывания апофермента с коферментом (чаще всего витамином или металлом). Кроме того, могут синтезироваться нестабильные легко распадающиеся молекулы ферментов. Все эти изменения структуры белков-ферментов называют молекулярными болезнями, или молекулярной патологией. Примерно 75% генных мутаций, ведущих к развитию Ф., представляют собой замену оснований в молекуле ДНК, что приводит к изменению генетического кода и соответственно к замене одной аминокислоты на другую в полипептидной цепи фермента. По принципу ведущих нарушений обмена веществ наследственные Ф. разделяют на следующие типы: · ферментопатии обмена аминокислот (алкаптонурия, альбинизм, гипервалинемия, гистидинемия, гомоцистинурия, гиперлизинемия, лейциноз, тирозиноз, фенилкетонурия, цистатионинурия, цистиноз); · обмена углеводов (галактоземия, гликогенозы, лактат-ацидоз, непереносимость фруктозы); · обмена липидов (липидозы) — плазматические (наследственная гиперлипидемия, гиперхолестеринемия, недостаточность лецитин-холестеринацилтрансферазы) и клеточные (ганглиозидозы, муколипидозы, сфингомиелинозы, цереброзидозы); · обмена пуринов и пиримидинов (подагра, синдром Леша — Найхана, оротовая ацидурия); · биосинтеза кортикостероидов (адреногенитальный синдром, гипоальдостеронизм); · обмена металлов — гепатоцеребральная дистрофия и болезнь Менкеса (обмен меди), гемохроматоз (обмен железа), семейный периодический паралич (обмен калия); · ферментопатии транспортных систем почек (тубулопатии) — почечный канальцевый ацидоз, болезнь де Тони — Дебре — Фанкони, фосфат-диабет (см. Рахитоподобные болезни), · ферментопатии желудочно-кишечного тракта — мальабсорбции синдром при недостаточности дисахаридаз, патология кишечного транспорта глюкозы и галактозы, врожденная хлоридная диарея. · нейромышечные (миопатии), · эндокринные, · печеночные, · кишечные,
60 наследственных болезней человека. Хромосомные болезни. Синдромы, связанные с нарушением плоидности, изменениями числа хромосом или нарушением их структуры: Патау, Дауна, Шерешевского – Тернера, Клайнфельтера и др.
К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Причиной хромосомных мутаций является нарушение структуры хромосомы под действием мутагенных факторов. Аномалии числа хромосом Болезни, обусловленные нарушением числа аутосом: Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала по 21-й хромосоме, либо полностью (трисомия), либо частично (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от степени копии, генетической истории и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например был обнаружен у обезьян и мышей). Совсем недавно исследователи вывели трансгенных мышей с наличием 21-й человеческой хромосомы (в дополнение к стандартному набору мышей). Добавление генетического материала может проводиться в разных направлениях. Типичный человеческий кариотип обозначается как 46,XY (мужской) или 46,XX (женский) (различие в поле несёт Y-хромосома). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2018-04-12; просмотров: 437. stydopedya.ru не претендует на авторское право материалов, которые вылажены, но предоставляет бесплатный доступ к ним. В случае нарушения авторского права или персональных данных напишите сюда... |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||